

Abstract
Fetal growth retardation is a fetal adaptation in response to inadequate supply of oxygen and/or nutrients. Animal models of intrauterine growth retardation are an invaluable tool to question the genetic, molecularand cellular events that determine fetal growth and development.Rodent and non-litter bearing animals are mammalian system with similar embryology,anatomy and physiology to humans. Utilization of these systems has led to a greater understanding of the pathophysiology and consequences of intrauterine growth retardation. These observations are comparable to that observed in humans born small for gestational age, and are of interest because of the known association between poor fetal growth and development of adult disease. Allthe experimental manipulations described here have altered a number of metabolic and physiological variables, but the pattern of alterations seems to vary with the procedure and species employed. This review describes animal models for intrauterine growth retardation and assesses their potentials and limitations at aiming to improve strategies for the prevention of adult disease.
Background
Type 2 diabetes mellitus (T2DM) and the Metabolic Syndrome are modern day plagues of societies in industrialized and developing nations alike [1]. The incidence of both of these diseases has reached epidemic proportions. Cardiovascular disease (CVD) is the leading cause of death in individuals with T2DM. The Metabolic Syndrome is characterized by any two of the following indicators: dyslipidemia, insulin resistance, obesity, impaired glucose tolerance [2]. While animal models [3, 4] (tables 1 and 2), and human epidemiologic studies [5, 6] illustrate that genetic and environmental factors play contributory roles in disease incidence and mortality, the underlying molecular mechanisms have been poorly defined. Fetal undernutrition and low birth weight (intrauterine growth restriction [IUGR]) [7,8,9] have been identified as risk factors for increased incidence of CVD, T2DM or their precursors such as dyslipidemia, impaired glucose tolerance or vascular endothelial dysfunction. Even after controlling for current body mass index, the risk for T2DM has been shown to be elevated among adults with low birth weights, suggesting that fetal programing confers risks above any subsequent environmental influences [10].
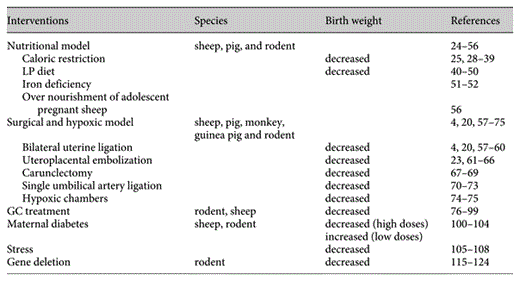

The development of disease in adulthood as a consequence of fetal programing was first described by Hales et al. [7,8,9] in the ‘thrifty phenotype’ hypothesis. This states that when maternal food restriction occurs, changes ensuring energy storage in the fetus take precedence. These changes may have conferred advantages on the fetus but result in disease after birth such as altered metabolic regulation leading to insulin resistance. Some have argued that the genetic variants may explain the association between reduced birth weight and increased risk of T2DM, thus ‘thrifty genotype’ is the real cause of adult disease [11].
An extension of the thrifty phenotype hypothesis is the ‘Predictive Adaptive Response’ theory which suggests that the fetus makes adaptations in utero or in an early developmental period based on the predicted postnatal environment [6]. How different the in utero environment is from the postnatal one is an important determinant of disease. The fetal adaptations that occur have been described as metabolic imprinting or programing [12]. This programing occurs within a specific developmental time period when a stimulus or insult results in abnormal development of a somatic structure or the inappropriate ‘setting’ of a physiological system leading to imbalances in adulthood.
Thus, based on evidence generated over 40 years early nutrition and growth affects CVD in adulthood, but is the prenatal window of growth the only critical period that will develop long-term disease? Increased infant growth rate by an enriched diet for a few weeks postnatally has been shown in observational as well as experimental studies to have a significant effect on CVD [13]. The effect of postnatal growth on cardiovascular complications has been demonstrated by alterations in systolic blood pressure [14], cholesterol [15] and insulin levels [16,17,18]. Thus, by limiting the consequences of adult disease to prenatal growth, we may be limiting ourselves to a vulnerable period (prenatal) in which intervention may be difficult to achieve. Instead, postnatal factors might be more amenable for targeting interventions making this critical period of growth a possible major contributor to long-term cardiovascular risk. Experimental animal models of IUGR will answer many of these questions.
This review will critically examine the experimental models in use to study IUGR and highlight their respective strengths and weakness.
Underlying Mechanisms
Animal Species Commonly Used in Models of Intrauterine Growth Retardation
Various animal species have been used as model of fetal growth retardation. One of the first animal models described by Wigglesworth [20] in 1964 was induction of uteroplacental insufficiency by uterine artery ligation in rodents, followed by the use of cigarette smoke during pregnancy in rats in 1969 [21], and experimental placental insufficiency in the rhesus monkey in 1971 [22]. Today, there are more than 1,500 publications using various animal models. Three quarters of them have been performed in rats (60%) and mice (10%), followed by sheep (12%), pigs (8%), guinea pigs (4%), monkeys (2%), chick embryos (2%), and lambs (2%). (The search phrase in Medline ‘animal name + fetal growth retardation’, search performed in May 2006.)
Many features of human biology at the cellular and molecular levels are shared with a wide variety of organisms. As mammals, the mouse and rat are highly related to humans, with similar genes, biochemical pathways, organs, and physiology. One of the major limitations is that rodents are altricial animals born with an underdeveloped brain and endocrine/paracrine system with significant maturation of organs during the weaning period. Furthermore, litter-bearing animal models (bearing multiple fetuses) may have a variable nutrient supply among the fetuses from the same litter. The pregnant sheep is a long-standing model for the study of placental–fetal interactions, and fetal growth restriction can be induced in pregnant sheep by maternal nutrient restriction, maternal nutrient excess, administration of glucocorticoids (GC), utero-placental embolization, carunclectomy (excision of uterine epithelium or caruncles) and maternal hyperthermia. Although all of these sheep models are capable of inducing fetal growth restriction, the degree of restriction is variable depending on the breed, maternal nutrition, and the intervention used [23]. Primates are the ideal animal model, but expensive housing, lifespan and ethical considerations limit their use. Thus, careful choice of an animal model is a critical step in the design of any study attempting to answer relevant questions associated with fetal programing.
Interventions
Nutritional Models
For over 30 years, nutrient manipulation during pregnancy has been an established model of growth restriction in humans and animal models [24, 25]. Nutritional intervention may mimic the challenge faced in underdeveloped countries. Different strategies have been used – global nutrient restriction, isocaloric low-protein (LP) diet, low-iron deficiency and overnutrition at different points during pregnancy. These models are based on the observation that fetal nutrient supply is one of the most important environmental factors affecting pregnancy outcome.
After the nutritional intervention, the animals are fed mainly a standard chow. Some commercially available diets incorporate soy (phytoestrogens) as the source of protein. These phytoestrogens (genestein and diadzein) may have beneficial actions [26, 27], lessen the development of CVD programed in utero. Thus, it is feasible that a weaning diet rich in phytoestrogens could reverse or decrease the insult caused by the different nutrient interventions during pregnancy.
Caloric Restriction
A number of studies have addressed the timing and effect of severe (70% of ad libitum), moderate (50%) and mild caloric restriction (30%) in various species. The most common dietary intervention has been a 50% reduction in caloric intake.
In rodents, caloric restriction from days 15 to 21 or from 7 to 21 during pregnancy does not affect length of gestation, litter size or mortality rate but significantly decreases birth weight [28,29,30,31,32,33]. In contrast, caloric manipulation from 7 to 14 days of gestation does not affect offspring birth weight [25]. Postnatal changes in body weight, body composition and food intake have been associated with both pre- and postnatal diet. Only mild and moderate caloric restriction in utero has been associated with increased catch-up growth postnatally. Obesity and hyperleptinemia have been associated with crossfostering (foster by a healthy mother), overfeeding and postnatal high-fat diet [25].
Caloric restriction programs hypertension, alteration in endothelial vasodilatation, decreased β-cell mass, islet number and insulin content, decreased insulin response to an oral glucose tolerance test, and hepatic insulin resistance [25].
Relatively few studies have addressed the impact of maternal undernutrition in non-litter-bearing species. The impact of poor fetal nutrition in sheep has been related to the timing of the insult. Maternal malnutrition from early to mid-gestation in fetal sheep leads to decreased maternal weight, fetal growth retardation and cardiac ventricular hypertrophy [34, 35], hypertension [36, 37], and blunted endothelial vasodilatation [38]. Similarly, in guinea pigs mild (85% ad libitum) or moderate (70% ad libitum) maternal food restriction reduces fetal growth and causes hyperinsulinemia in young adult male offspring [39].
Low-Protein Diet
Initially described by Snoeck et al. [40] pregnant rats are fed either a normal diet (20% protein) or an isocaloric LP diet (8% protein), from the first day to until the end of gestation. Protein restriction adversely affects fertilization as well as early egg development and differentiation in mice [41], and alters placental transfer of amino acids in guinea pig [42]. At mid-pregnancy, glucose, insulin, prolactin and progesterone levels are significantly lower in LP mothers compared to control [43]. Litter size does not differ between groups, but LP pups are born IUGR compared to the control.
One advantage of a LP diet is that it can be done in various animal models. LP diet consistently programs hypertension [44], pancreas development, altered insulin response to an oral glucose tolerance test, hepatic insulin resistance and in non-litter-bearing species has been shown to program endothelial dysfunction [45].
One of the major differences between the LP diets is that the composition of the diet elicits different programing effects. Two different LP diets have been commonly used in animal models of fetal programing: The Southampton and the Hope farm diet. The Southampton diet programs hypertension [44], while the Hope farm diet programs insulin resistance [46]. The difference between these diets is the balance of specific amino acids, fat and carbohydrate used. The Southampton diet [44] differs from the Hope farm diet [46, 47] in the oil composition (soybean vs. corn oil), methionine and carbohydrate content. Amino acids like methionine and glycine are essential compounds for a number of metabolic functions during development such as nucleic acid, collagen and heme formation. Methionine is a major component of the Southampton diet, leading to an excess in comparison to other amino acids. Methionine excess causes hyperhomocystinemia, a known cardiovascular risk factor for the development of endothelial dysfunction. Interestingly, the addition of glycine [48, 49] as well as folate reverses the hyperhomocystinemia in animals fed the Southampton diet further suggesting that methionine excess may in part explain the fetal programing observed in this model. Hope farm diet has three times more of the ω-3 polysaturated (PUFA) fatty α-linoleic acid than Southampton diet adding a possible cardioprotective advantage. The carbohydrate content should be also assessed because a high glycemic index (Hope farm) may program a different phenotype in addition to the protein restriction. Sodium content is another parameter to be considered before choosing a nutrient modification. Inappropriate sodium intake during pregnancy has been associated with programing of high blood pressure and CVD [50].
Iron Deficiency
The association between anemia in pregnancy and low birth weight [51] has prompted groups to study the relationship between maternal iron status and the development of adult disease. Animal are fed either a control diet (50 mg Fe (kg diet)–1) or a Fe-deficient diet (7.5 mg Fe (kg diet)–1)for 4 weeks before mating and through different stages of gestation. Iron deficiency has no effect on maternal weight or litter size, but body weight at birth decreases withthe severity of the deficiency [52].
Over-Nourishment of Adolescent Pregnant Sheep
In Pima Indians the association of birth weight and diabetes is U shaped, with the highest prevalence of diabetes occurring in both high (LGA) and low birth weight infants [53]. Pre-pregnancy obesity seems to be independentlyand strongly associated with LGA while net weight gain (5 kg) during pregnancy had a more modest [54], little or no effect on birth weight [55].
Overfeeding single pregnant adolescent dams to promote rapid maternal growth throughout pregnancy is associated with prematurity and poor placenta/fetal growth. The experimental model consists of singleton pubertal adolescent ewes pregnancies that are fed twice their energy requirements. Pregnancies are characterized by placental insufficiency resulting in fetal hypoxia, hypoglycemia, hypoinsulinemia and reduced fetal growth [56].
•Nutrient manipulation during pregnancy, as a cause of IUGR, does not cause frank diabetes, although it does cause CVD.
•Some of the weaknesses of these models are the variability of the outcomes that are in part dependent on the species studied, time of the insult, maternal metabolic environment and body composition before pregnancy.
•The balance of essential amino acids and other nutrients may be critical in determining the metabolic outcome of the offspring.
Surgical and Hypoxic Models
Surgical interventions have been used to generate a hypoxic model in which growth restriction will affect the placenta and the fetus. Such models are highly relevant to human pregnancy in developed countries. Low oxygen concentrations will generate: fetal hypoxia, increase or decrease amino acid levels, hypoinsulinemia and altered organ growth, factors that cannot be dissociated from each other.
Bilateral Uterine Ligation
Three-month-old pregnant rats on day 19 of gestational age (term 21.5 days) undergo bilateral uterine artery ligation [20]. Suture is placed around both uterine arteries, then either tied or withdrawn before closing the abdomen. Thirty percent of the fetuses die or undergo partial resorption, especially at the vaginal portion of the horn. Fetuses from bilateral uterine ligation show a 20% lower body weight compared to control, and the body weights remain significantly diminished even at 21 days of life. Litter size does not differ between groups. Similar to humans, fetal rats in this model suffer transient hypoglycemia, hypoxia, and acidosis, and demonstrate the long-term effects of the altered intrauterine milieu [57, 58]. These animals exhibit mild insulin resistance and β-cell secretory defects very early in life [4, 59]. Eventually, the β-cell secretory capacity fails and overt diabetes occurs. A similar model has been used in mice in which fetal growth retardation is induced on 14 day of pregnancy, resulting in a reduction in fetal growth [60].
Utero-Placental Embolization
The pregnant sheep is a long-standing model for placental–fetal interactions, and fetal growth restriction can be induced by utero-placental embolization, carunclectomy or maternal hyperthermia [23]. In sheep, fetal hypoxia (50% of control) has been induced by umbilico-placental embolization, from 120 days of gestation until term [61, 62]. Embolization is produced by injection of microspheres of 15–30 µm in diameter into the fetal or maternal placental vascular bed producing fetal hypoxemia, temporary increase of fetal cortisol and norepinephrine levels [63,64,65,66], and growth retardation. During pregnancy IUGR fetuses have lower blood glucose and at birth lambs weigh 33% less than controls [62].
Carunclectomy
Carunclectomy is the excision of all visible uterine epithelium caruncles (dark spots in utero) that will determine the number of cotyledons that will generate the placental mass [67]. Placental and fetal weight varies among pregnancy, and fetuses are hypoxemic and hypoglycemic during gestation [67,68,69]. Thus, it is the small size of the placenta per se rather than alterations in its nutrient transfer or capacity and or hormonal milieu that is the major limitation to fetal growth in the growing adolescent sheep.
Single Umbilical Artery Ligation
Chronic fetal hypoxia and IUGR can also be experimentally induced by single umbilical artery ligation in catheterized fetal sheep at 105–110 days of gestational age (term mean 147 days). Catheterization causes preterm labor, hypercortisolism and growth retardation [70].
Unilateral artery ligation has also been used in guinea pigs as the preferred method to provide placental insufficiency. The procedure is done at mid-gestation (at 28–30 days, term 67 days) causing significant fetal death. The surviving fetuses have either similar or 60% decreased birth weight [71, 72]. IUGR fetuses are hypoglycemic, hypoinsulinemic, hypocortisolemic with elevated glucagon [72], insulin growth factor-binding globulin (IGFBP)-2 and -4, and reduced IGFBP-3 levels [73].
Hypoxic Chambers
Another model of hypoxia is the acute or chronic exposure of pregnant rats to lower oxygen concentration (9.5–14% vs. 21% in controls) in specially designed chambers. Independently of the duration of the hypoxic insult, genes related to calcium homeostasis and cytoskeleton are consistently up-regulated and genes related to G-protein signaling, growth, and ion transport are consistently down-regulated in all tissues. Acute hypoxia is associated with up-regulation of hexokinase, phosphofructokinase, and aldolase while chronic hypoxia is associated with up-regulation of inflammation-related genes [74]. Fetuses have lower body weight compared to controls. Lower oxygen concentration or chronic hypoxia results in decreased daily food intake (similar to models of caloric restriction), litter size and severe growth retardation [74, 75].
•A major advantage of the surgical model is that the maternal metabolic environment and body composition before and during pregnancy will not affect the fetal outcome.
•Surgical manipulations are particularly useful in large animal models, such as sheep, since the outcome can be monitored in a viable fetus. In small animals 30% of the fetus either die or undergo partial resorption.
•Bilateral uterine artery ligation provides the only model that develops frank diabetes, due to the association between insulin resistance and β-cell secretory defects.
•It is possible that surgical manipulations could be used in conjunction with the other models to further elucidate the effect of the maternal versus the placental milieu on fetal growth.
Glucocorticoids Model
Maternal exposure to GC has been associated with reduced birth weight and adult disease in both humans and animals models [76,77,78,79,80,81]. In humans, the effects of prenatal GC exposure are not clear. Impaired fetal growth was not identified in the original randomized trialsof prenatal GC given for fetal lung maturation [82]. Other studies have demonstrated an association between repeated administration of antenatal corticosteroidsand impaired fetal growth in preterm [83,84,85] and term infants [86]. Exposure to multiple courses of antenatal corticosteroids compared with a single course resulted in a reduction in birth head circumference with no alteration in birth weight [87]. Thus, the National Institutes of Health (NIH) published in 2000 a consensus statement that reaffirmed the safety and efficacy of a single course of antenatal steroids, but emphasized that there are no data to support the safety and efficacy of repeated courses.
In animals, the effect on birth weight is more notable with increasing number of GC courses [84, 88] or when given in the latter stages of pregnancy [76]. The exposure to natural (cortisol) or synthetic GC (dexamethasone or betamethasone) does not necessarily have the same long-term consequences in the offspring [89]. Dexamethasone treatment but not cortisone acetate (not the natural GC of rodents) or betamethasone causes hypertension in the adult offspring. Furthermore, the long-term effect of GC may vary among species or sex [90]. In adult female the hypothalamo-lactotroph axis is profoundly affected suggesting a sex-specific effect by perinatal exposure to GC [90]. Maternal administration of betamethasone, but not cortisol, causes decreased fetal growth in lambs [91]. In long-gestation mammals (sheep or humans) the timing of the insult is more important than the type of GC excess [92]. Early exposure of GC in the ewe has been associated with adult hypertension [93,94,95]. In contrast, late exposure during gestation to GC is associated with insulin resistance but not hypertension [79]. Interestingly, excessive prenatal GC exposure may also be a result of caloric/protein restriction models, suggesting that mothers who are stressed during a particular stage of pregnancy may predispose the offspring to adult disease [92]. Some of the protocol in use are: intraperitoneal injections of dexamethasone (0.2 mg/kg body weight), or via the mothers’ drinking water (1 µg/ml) to pregnant rats from gestational day 11 [90, 96], or betamethasone (50–600 µg/kg per day, subcutaneously) from gestational day 15–21 [97], or hydrocortisone acetate (1.5 mg/day/animal, subcutaneously) from day 17 to 19 of pregnancy [98]. Similarly to corticosteroids, prenatal bi-weekly testosterone treatment (100 mg) to pregnant sheepbetween 30 and 90 days of gestation produces growth-retarded offspring [99].
•The impact of exposure of the fetus to GC depends on: dose and type of GC, window of exposure, sex and stage of development of the developing fetus, and animal model, suggesting that part of differences in the outcome may be related to the critical window of development of each organ, species and sex. Whether this is the case in humans awaits long-term follow-up of children enrolled in randomized controlled trials of prenatal GC therapy.
Maternal Diabetes
Uncontrolled maternal diabetes is characterized by an abnormal intrauterine milieu that results in alterations in growth and development of the fetus [100,101,102,103,104].
The dose of streptozotocin (STZ) may be critical in determining the metabolic outcome of the offspring.
•Diabetes is induced by administration of STZ to either rats or sheep. This method has been used to elucidate the effect of diabetes on fetal growth and development. A major drawback is that the phenotype of the offspring is related to the dose of STZ used during pregnancy. Low dose of STZ results in mild gestational diabetes and fetal macrosomia, whereas a high dose induces insulin deficiency diabetes-associated growth restriction.
Stress-Induced Fetal Growth Restriction
Stress interferes with reproduction, and has been demonstrated to adversely influence fetal growth. Maternal stress has been induced by placement of pregnant females for 45 min in a transparent plastic cylinder three times per day [105], or by exposure to sound (100 dB, random frequency between 9 and 34 kHz) for 8 h three times during pregnancy, or by housing for ∼85 consecutive days in a heat chamber in which the temperature reached 40°C [106], or by maternal restraint [107, 108].
Other Maternal Models
Fetal alcohol exposure causes reduced birth weight. Animal models of fetal alcohol growth restriction include inhalation of ethanol vapor and intraperitoneal injection, as well as oral exposure by gavage, diet, or drinking water [109]. Inhalation of tobacco on days 3–22 of gestational age [21], renal insufficiency [110], partial nephrectomy on day 14 of gestational age [111], and maternal thyroidectomy [112].
Fetal and Genetic Interventions
Several manipulations at the level of the fetus have been described as possible models for growth restriction like transplacental infection at day 12.5 of gestation by direct injection of cytomegalovirus [113]; or pancreatectomy of sheep fetuses [114]. Gene deletions in mouse models have also been associated with IUGR, e.g. pleomorphic adenoma gene 1 proto-oncogene [115], insulin receptor substrate-1 [116], placental specific knockout of a single imprinted gene encoding insulin-like growth factor (IGF)-2 [117], total ablation of the glucagon receptor [118], ablation of the scaffold attachment factor B1 [119], testicular orphan nuclear receptor 4 [120], IGF-2 mRNA-binding protein 1 [121], insulin receptor and insulin gene, and IGF-1 and -2 [122], IGF-1 receptor [123], and IGFBP-1 overexpression [124].
Conclusions
In conclusion, animal models of growth restriction are an effective and valuable tool in understanding the relationship between fetal growth and adult disease. All the experimental models have produced useful information into the basic mechanism underlying fetal growth, and are necessary for the advancement of the field as well as to prevent the development of disease. While we cannot substitute for the study of human disease, animal models of growth restriction provide a useful tool that allows the utilization of techniques that would be unethical in humans. The caveat of in vivoresearch lies in the observation that the majority of animal models manifest most but not all the symptoms seen in humans; frank disease is not easily reproducible. A possible explanation is that the critical window for programing is different among the species. Further, the maternal hormonal milieu, placental function and fetus response to the insult may vary among species and techniques used to generate growth restriction. Some have argued that the ‘thrifty phenotype’ may be due to a ‘thrifty genotype’ and that genetic variants may explain the association between reduced birth weight and T2DM, suggesting that programing is more a risk factor than a causal event in the history of T2DM. Thus, as the developmental origin of adult disease field expands we need to balance the contribution of the thrifty phenotype and the thrifty genotype in the generation of disease, and determine their respective role in the different outcomes. Therefore, we may conclude that though, none of the models currently available are totally satisfactory, because of the possible confounding variables that may play a role in the pathogenesis of fetal growth restriction (e.g. genetic vs. environment), the challenge is to fully understand in detail the specific mechanism that leads to metabolic disease in the offspring.
Acknowledgement
Dr. Vuguin is a recipient of the National Institute of Health KO8 HD042172 award.
