

Abstract
Tissue hypoxia occurs when local metabolism is disturbed by an imbalance between oxygen supply and consumption. In patients with chronic kidney disease, chronic hypoxia in the kidneys is the end result of multiple processes and mechanisms. Once established, however, accumulating evidence points to this chronic hypoxia as the central player and final common pathway to end-stage renal disease. The cellular response to hypoxia is centered on hypoxia-inducible factor, HIF. This factor is composed of two subunits, an oxygen-sensitive HIF-α subunit and a constitutively expressed HIF-β subunit. Intracellular accumulation of HIF induces the coordinated expression of a number of adaptive genes against hypoxic insult.Three isoforms of HIF-α subunits have been identified, HIF-1α, HIF-2α, and HIF-3α, of which HIF-2α is involved in the regulation of erythropoietin as well as oxidative stress. HIF is regulated by prolyl hydroxylation and asparaginyl hydroxylation of the HIF-α subunit at the protein level. Because HIF is activated only to suboptimal levels in various pathogenic states, therapeutic activation holds promise as a novel and effective approach to the future care of end-stage renal disease.
Oxygen, an Essential Molecule for Life
Virtually all organs are dependent on a sufficient and consistent supply of oxygen. Oxygen is essential to many metabolic processes, including oxidativephosphorylation, in which it serves as an electron acceptorduring ATP formation. This energy production system is in fact so efficient that it yields a total of 38 ATP molecules per mole of glucose oxidized, in contrast to anaerobic glycolysis, which produces only 2. In fact, the overall environmental increase in oxygen during the past 200 million years has played a critical role in the evolution of placental mammals: oxygen exchange via the placenta forces the fetus to live at a low arterial oxygen pressure, and successful reproduction accordingly requires relatively high ambient oxygen concentrations [1].
Hypoxia, the Final Common Pathway to End-Stage Renal Disease
Although blood flow to the kidney is relatively high, the presence of oxygen shunt diffusion between arterial and venous vessels that run in close parallel approximation keeps renal tissue oxygen tension comparatively low. Deprivation of oxygen as an essential metabolic fuel thus serves as an establishedcommon final pathway to end-stage renal disease. This mechanism was originally proposed by Norman and Fine as the chronic hypoxia hypothesis [2], and has been validated by a large number of studies since (for recent reviews, see table 1). With regard to cellular location, the final common pathway of chronic hypoxia operates principally in the tubulointerstitium.

The development of chronic hypoxia in the tubulointerstitium actually occurs via multiple mechanisms (fig. 1). Glomerular efferent arterioles enter the peritubular capillary plexus, which surrounds the tubules and offers oxygen to tubular and interstitial cells. Extensive tubulointerstitial injury is associated with damage to the renal arterioles and arteries as well as with distortion and loss of peritubular capillaries, leading to a decrease in peritubular capillary flow and oxygen supply. Moreover, by distancing capillaries from the tubular cells, the subsequent interstitial fibrosis reduces oxygen diffusion efficiency, thereby impairing tubular oxygen supply even further.

Multiple mechanisms of chronic hypoxia in the kidney. Various factors induce hypoxia of the kidney via both structural and functional changes.
Multiple mechanisms of chronic hypoxia in the kidney. Various factors induce hypoxia of the kidney via both structural and functional changes.
A number of other mechanisms induce tubulointerstitial hypoxia even before the development of fibrosis. Glomerular injury and vasoconstriction of arterioles due to imbalances in vasoactive substances such as angiotensin II and nitric oxide functionally decrease postglomerular peritubular capillary blood flow, while oxidative stress and dysregulation of hepatic arginine metabolism impair oxygen utilization efficiency by exaggerating mitochondrial respiration, and thereby induce relative hypoxia in the kidney [3]. The issue of enhanced transport burden in remnant nephrons is also crucial. In order to compensate for the loss of nephrons, remnant nephrons need to deal with enhanced transport burden. Because of increased demand for energy required for active transport, oxygen consumption is increased in remnant nephrons, resulting in a decrease in local oxygen tensions. Further, renal anemia hinders oxygen delivery.
Together, these mechanisms act at various points in the disease course to result in chronic hypoxia of the kidney.
Hypoxia-Inducible Factor as a ‘Master Gene’ Switch to Protect Organs against Hypoxia
Against this multitude of threats, organisms have developed an efficient system to protect tissues suffering from hypoxia.
To economize energy use under hypoxic conditions, cells suppress protein synthesis. They do this by regulating the initiation step of mRNA translation, as follows. First, hypoxia activates the unfolded protein response, which occurs in response to endoplasmic reticulum stress. The subsequent phosphorylation of eukaryotic initiation factor 2α inhibits the translation of most genes. However, at the same time as this pathway is bringing about a general inhibition of translation, the cell is also activating other pathways essential to survival, which results in the expression of genes involved in adaptation to hypoxia.
Importantly, this multi-actor response occurs via the activation of a ‘master gene’ switch that results in a broad and coordinated downstream reaction. Sitting at the center of this web of cellular responses to hypoxia is hypoxia-inducible factor, HIF.
Structurally, HIF is composed of an O2-regulated α-subunit and a constitutively expressed β-subunit (arylhydrocarbon receptor nuclear translocator, ARNT), both of which belong to the basic helix-loop-helix (bHLH)-PAS (PER, ARNT, SIM) protein family. Transactivation involves dimerization of the two HIF subunits, which then binds to an enhancer element called the hypoxia-responsive element in target genes.
HIF is constitutively transcribed andtranslated. Its production is primarily regulated by the rate of degradation. The HIF-α subunit interacts with a protein that regulates its half-life, namely von Hippel-Lindau tumor suppressor protein (pVHL). This interaction is triggered through HIF-α hydroxylation, which in turn is catalyzed by a set of oxygen-dependent enzymes. This interaction is triggered through HIF-α hydroxylation, which in turn is catalyzed by a set of oxygen-dependent enzymes which cause the hydroxylation of specific proline residues within the oxygen-dependent-degradation domain by specific HIF-prolyl hydroxylase domain proteins (PHDs; fig. 2). The hydroxylated protein is then recognized by pVHL, which functions as an E3 ubiquitin ligase. Thus, under normoxic conditions, HIF-α is targeted for ubiquitinylation by pVHL and rapidly degraded by the proteasome. Concurrently, the asparagine residue within the COOH-terminal activation domain (C-TAD) is also hydroxylated by an asparaginyl hydroxylase (also referred to as FIH-1), which prevents the coactivator p300/CBP from binding to the HIF-α subunit.
![Fig. 2. Structure of the HIF-α subunit. HIF-1α and HIF-2α are structurally similar. Hydroxylation of two proline residues by PHD earmarks the protein for degradation, whereas hydroxylation of an asparagine residue by FIH reduces its transcriptional activity. HIF-α possesses two transcriptional activation domains [TADs; NH(2)-terminal (N-TAD) and COOH-terminal (C-TAD)], and both the N-TAD and C-TAD are important for optimal HIF transcriptional activity. N-TAD overlaps with oxygen-dependent-degradation domain (ODDD).](https://karger.silverchair-cdn.com/karger/content_public/journal/nee/110/1/10.1159_000148256/1/m_000148256_f02.gif?Expires=1716291489&Signature=VQ66tcEMjEiS3qiAA43wxmzR2u-48xjeNz1r6rngQ5wkrzbo41p7tOOlxHvt04nDvmwvH3IWILvJX9Oma3zsX2HkyF4NaUGCLEKQMjRwPDiM0UELDbFg-V-FQnS7YIuhezv6vW-5qFLCWKB9zQdGGKJKtcB9eyH-ZGeT4NUp7KGKPNUchQdKVuxbNXGtuXMzKYgwKCzScvGtLV7Mz8YxE9hwaD3dXKKG6WdqeNqVR1Da01frre6Q-~Q00M2kGoAVMbxsM9h-vadYbCsCntdOKFsvtWNQeCwKTeFgvQC6fCH3BwZuF0wUTmMs3umwrkiZXSl~c9IfHuFBAn7j5~JSpA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Structure of the HIF-α subunit. HIF-1α and HIF-2α are structurally similar. Hydroxylation of two proline residues by PHD earmarks the protein for degradation, whereas hydroxylation of an asparagine residue by FIH reduces its transcriptional activity. HIF-α possesses two transcriptional activation domains [TADs; NH(2)-terminal (N-TAD) and COOH-terminal (C-TAD)], and both the N-TAD and C-TAD are important for optimal HIF transcriptional activity. N-TAD overlaps with oxygen-dependent-degradation domain (ODDD).
Structure of the HIF-α subunit. HIF-1α and HIF-2α are structurally similar. Hydroxylation of two proline residues by PHD earmarks the protein for degradation, whereas hydroxylation of an asparagine residue by FIH reduces its transcriptional activity. HIF-α possesses two transcriptional activation domains [TADs; NH(2)-terminal (N-TAD) and COOH-terminal (C-TAD)], and both the N-TAD and C-TAD are important for optimal HIF transcriptional activity. N-TAD overlaps with oxygen-dependent-degradation domain (ODDD).
PHDs function with lowefficiency under conditions of hypoxia, and nonhydroxylated HIF-α cannot interact with pVHL. Stabilized HIF-α then binds to its heterodimeric partner HIF-β,and together these proteins act in the nucleus to transactivategenes involved in adaptation to hypoxic-ischemic stress.Although three HIF prolyl hydroxylases with the potential to catalyze this reaction have been identified, termed PHD1, PHD2 and PHD3, the relative importance of the individual PHD isozymes and FIH in the hypoxic response in different cell types has yet to be fully defined.
Isoforms of HIF
The activated HIF then proceeds to drive the transcription of more than 70 genes, which variously participate in erythrocytosis, angiogenesis, glucose metabolism, and cell proliferation/survival, to bring about the many compensatory responses to oxygen deprivation at both cellular and physiological levels.
Three isoforms of the HIF-α subunit have been identified, HIF-1α, HIF-2α, and HIF-3α. HIF-1α and HIF-2α are structurally and functionally similar and share 48% overall amino acid identity. In contrast, while HIF-3α has high similarity with HIF-1α and -2α in the bHLH and PAS domains, it lacks structures for transactivation found in the C-termini of HIF-1α and -2α, suggesting that it may be a negative regulator of hypoxia-inducible gene expression.
While most cell types express HIF-1α, HIF-2α shows a more restricted pattern of expression, which includes the developing vasculature. In the kidney, HIF-1α is expressed in tubules, while HIF-2α is confined to endothelial and interstitial cells. Knockout of either results in embryonic or perinatal lethality, suggesting a nonredundant role for both.
Recent molecular genetics studies in mice have identified HIF-2α as a physiological regulator of erythropoietin (table 2). In humans, a mutation of HIF-2α which stabilizes the product was shown to be a cause of familial erythrocytosis [4]. In polycystic kidney disease, cyst expansion may result in pericystic hypoxia and consecutive upregulation of pericystic HIF-2 induction, leading to comparatively high hemoglobin concentrations [5].

In addition to erythropoiesis, HIF-2 plays a crucial role in defense against oxidative stress. Although the notion of oxidative stress under hypoxic conditions sounds paradoxical, hypoxic cells do in fact suffer from energy depletion and oxidative stress (fig. 3). Knockout of PHD1 in mice reduces the occurrence of oxidative stress with subsequent lowering of oxygen consumption in skeletal muscle. This metabolic adaptation to oxygen conservation, which provides acute protection of myofibers against lethal ischemia, relies primarily on HIF-2 [6]. Studies in HIF-2 knockdown mice show that HIF-2 in the endothelium exerts a protective effect against ischemia in the kidney by ameliorating oxidative stress [7].
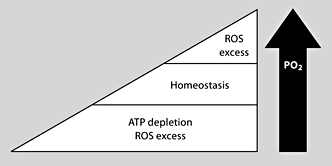
Oxygen tension and cellular homeostasis. Both an increase and a decrease in oxygen tension lead to oxidative stress. ROS = Reactive oxygen species.
Oxygen tension and cellular homeostasis. Both an increase and a decrease in oxygen tension lead to oxidative stress. ROS = Reactive oxygen species.
HIF Activation Is Not Enough in Kidney Disease
If HIF activation were already maximal under pathological conditions, therapeutic approaches which target HIF might be of little use. In kidney disease, however, this is not the case; HIF activation is suboptimal, and the door to this strategic route is accordingly wide open. Evidence for this comes from a variety of studies. HIF activation is suboptimal in diabetic conditions, and is augmented by treatment with antioxidants [8, 9]. HIF accumulation in acute ischemia was substantially less thanthat seen in animals exposed to carbon monoxide, indicating that activation was also submaximal in renal ischemia with complete shutdown of blood flow [10]. In a model of rhabdomyolysis, transcriptional hypoxia adaptation in the most affected tubules was transient and heterogeneous [11], while proteinuric states may also inhibit optimal HIF activation [12]. Rosenberger et al.[ 13] utilized an ex vivomodel of the isolated perfused rat kidney with controlled oxygen consumption and provided evidence for a ‘window of opportunity’ for HIF activation, under moderate sublethal hypoxia, whereas a more severe hypoxia results in suppression/disappearance of HIF and induction of apoptotic cell death. Together, these reports suggest the potential of pharmacologic enhancement of HIF to improve outcomes.
Activation of HIF Is Effective in Kidney Disease
Thanks to the powerful and coordinated response it produces against hypoxia, modulation of HIF activity should be effective in a variety of hypoxic states. This concept has been validated by gene transfer studies of constitutively active HIF into the rat kidney.
How might this best be achieved? The finding above that HIF level is determined by hydroxylation-induced degradation suggests that the hydroxylases in this reaction might be good targets for therapeutic intervention. PHD requires iron as a cofactor to hydroxylate the critical prolines onHIF-α, and some of the best-established activators of HIF are chelatorsof iron. Among the most well-established chelators in HIF activation are desferoxamine and cobalt chloride. Chemical preconditioning with cobalt chloride protected the kidney in an ischemia-reperfusion model of the kidney, while cobalt chloride improved disease manifestations in a variety of kidney disease models even when administration was started after the initial insult. Other stimuli which activate HIF have also shown renoprotective effects, including carbon monoxide (table 3).

Expectations for HIF activation in not only kidney disease but also conditions involving other organs such as the heart and brain have attracted much recent effort in the development of specific and nontoxic HIF activators. While screening a siRNA library against the entire druggable genome failed to identify suitable candidates, possibly due to off-target effects of siRNA, screening of a small-molecule library using an HIF reporter cell line revealed a class of alkyliminophenylacetate compounds which inhibit hypoxia-induced HIF reporter activity at single-digit nanomolar concentrations, possibly via the blockade of hypoxia-induced mitochondrial reactive oxygen species production [14]. This method may also be useful in identifying HIF activators. Employing a different strategy, namely docking simulation based on the 3-dimensional structure of PHD2, we identified novel HIF-activating compounds which induced angiogenesis in a mouse sponge assay and protected the brain against ischemic injury in gerbils [15]. These technological advances will help us to develop more specific HIF activators in years to come.
The Yin and Yang of HIF Activation
A fine line between therapeutic benefit and harmful side effects must always be drawn. Any novel use of HIF activation requires the careful consideration of overall net effect. Iron is a necessary cofactor fora host of important cellular functions, including oxidativephosphorylation and arachidonic acid signaling. We may find that thepotential side effects of iron chelation hinder its therapeutic use in activating HIF. Indeed, the pharmaceutical effect of HIF activation cannot be free from side effects. pVHL knockout mice showed de novo expression of the HIF target gene Cxcr4 in glomeruli in association with the development of rapidly progressive glomerulonephritis [16]. HIF activation enhances angiogenesis, and may thus be associated with a range of adverse effects, especially on systemic application in patients with known or as yet unrecognized coexistent conditions, such as the induction of occult tumor growth. One HIF target gene is connective tissue growth factor (CTGF), and while CTGF is an angiogenic factor, it also serves as a profibrotic factor, and inappropriate long-term HIF activation may thus lead to fibrosis [17].
Conclusion
Chronic hypoxia is the final common pathway to end-stage renal failure. Hypoxia of the kidney is induced by multiple mechanisms, and therapeutic approaches against this final common pathway should be effective in a broad range of renal diseases. HIF is the master switch of the many hypoxic adaptation responses. Our challenge is to investigate the different pathways of HIF regulation and discover novel HIF-affecting drugs. We are confident that the near future will witness new discoveries for HIF and HIF-regulated pathways that will enhance and facilitate strategies to protect the kidney.
Acknowledgement
The authors were supported by a Grant-in-Aid for Scientific Researchfrom the Japan Society for the Promotion of Science (19390228).
