

Abstract
Noonan syndrome (NS) is one of the most common syndromes transmitted by a mendelian mode. In recent years, germline mutations that affect components of the RAS-MAPK (mitogen-activated protein kinase) pathway were shown to be involved in the pathogenesis of NS and four rare syndromes with clinical features overlapping with NS: Leopard syndrome, cardio-facio-cutaneous syndrome, Costello syndrome and neurofibromatosis type 1. Several hormones act through receptors that stimulate the RAS-MAPK pathway, and therefore, NS and related disorders represent a remarkable opportunity to study the implication of the RAS-MAPK pathway in different endocrine systems. Additionally, children with NS frequently are referred to the endocrinologist because of short stature, delayed puberty and/or undescended testes in males. In this paper, we review the diagnostic, clinical and molecular aspects of NS and NS-related disorders.
Introduction
Noonan syndrome (NS; OMIM 163950) is one of the most common syndromes transmitted by a mendelian mode. The incidence of affected individuals is estimated to be between 1:1,000 and 1:2,500 [1, 2]. NS is a clinically heterogeneous disorder predominantly characterized by dysmorphic facial features (fig. 1), congenital heart disease (most commonly pulmonary valve stenosis, hypertrophic cardiomyopathy and atrial septal defects), proportionate post-natal short stature, neck abnormalities and chest deformities [1, 3, 4] (table 1). Mild mental retardation, bleeding diathesis, lymphedema, hearing difficulty and cryptorchidism are also occasionally observed in affected individuals. NS has an equal male to female ratio [5, 6]. Familial cases correspond to approximately 20% of the cases [1] and presented mostly an autosomal dominant inheritance with a near complete penetrance [7].
![Phenotypic abnormalities associated to NS [1, 3, 4]](https://karger.silverchair-cdn.com/karger/content_public/journal/hrp/71/4/10.1159_000201106/2/m_000201106_t01.gif?Expires=1716587543&Signature=T2NPVQ3AFDu7MWABYG6MVe6bbJLNezSab2cMOZyEznrNbDsuvN1UcnwQb7Z-JxyFoCOoCbLMaeauvaU12a8Z~bTzEVgQpMjfdlfU~q81zKWT8K0Eec0RNP7Av-SGZACQLKY2yoQY6WjvMRn6u3GJPxDLv3IuD5PIvj5l8blCCtK6BWB~OXGJ0Ysq0W7EClf9zBUr5ikFmSHls7dFZcmixWYo4LzwoANOq8-eq2pK2feI9xSEZmGmkit-QLp6TEXkIC2x3WMMHYmcVkpPUOK6DqBkEgvijRcuybrhxvFbSLW3c7d0yNxIgtRcyEetptEgez~QHbuY5gm34vnOIfYfNA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)

NS features: (a) eye abnormalities: ptosis, hypertelorism, epicanthal folds; (b) webbed neck, and (c) ear abnormalities: low-set posteriorly rotated ears and thick helix.
NS features: (a) eye abnormalities: ptosis, hypertelorism, epicanthal folds; (b) webbed neck, and (c) ear abnormalities: low-set posteriorly rotated ears and thick helix.
In recent years, germline mutations that affect components of the RAS-MAPK (mitogen-activated protein kinase) pathway were shown to be involved in the pathogenesis of NS and of four rare syndromes with NS overlapping features: Leopard syndrome (OMIM 151100), cardio-facio-cutaneous syndrome (CFC; OMIM 115150), Costello syndrome (OMIM 218040) and neurofibromatosis type 1 (NF1; OMIM 162200) [8].
Several hormones, including growth hormone (GH) and insulin-like growth factor 1 (IGF-1), act through receptors that stimulate the RAS-MAPK pathway, and therefore NS and related disorders represent a remarkable opportunity to study the implication of the RAS-MAPK pathway in different endocrine systems. Additionally, children with NS are frequently referred to endocrinologists because of short stature, delayed puberty and/or undescended testes in males. Therefore, NS is important in the differential diagnosis of short stature and has important implications in GH therapy [9,10,11].
Differential Diagnosis of Noonan Syndrome
Although NS girls can be misdiagnosed as Turner syndrome (TS) because of clinical feature similarities between these two syndromes, the presence of hypergonadotrophic hypogonadism and abnormal karyotype in TS girls facilitates the distinction between these two syndromes. Several other disorders, with normal gonadal function and normal karyotype, resemble NS phenotypically and constitute the true differential diagnosis of NS.
Leopard syndrome is an autosomal dominant disorder that shares some clinical characteristics with NS. The acronym refers to the major features: Lentigines, ECG conduction abnormalities, Ocular hypertelorism, Pulmonic stenosis, Abnormal genitalia, Retardation of growth and sensorineural Deafness [12, 13]. The presence of café-au-lait spots in early infancy and generalized multiple lentigines after 5–6 years of age are the main characteristics of Leopard syndrome (fig. 2).

In addition to Leopard syndrome, two other rare syndromes, Costello syndrome and CFC syndrome, present facial features and cardiac malformations that resemble NS. In comparison with NS, Costello and CFC syndromes present a coarser face and both syndromes are associated with more frequent and severe developmental delay. Costello syndrome patients usually have macrocephaly, cutis laxa, nasal and perioral papillomata, deep palmar and plantar creases, diffuse skin hyperpigmentation and nail dysmorphology, as well as an increased risk of malignancy, especially rhabdomysarcoma. CFC syndrome patients typically have ectodermal abnormalities such as sparse hair and eyebrows, follicular hyperkeratosis, palmoplantar hyperkeratosis and an ichthyosis-like condition [14, 15].
Clinical Diagnosis of Noonan Syndrome
Diagnosis of NS is primarily based on clinical findings. Generally, classical facial features or a typical cardiac malformation trigger suspicions of NS. In the newborn, facial features can be less apparent, but generalized edema, excess nuchal fold and congenital heart defect can suggest the diagnosis.
Widely variable facial appearance is observed among NS patients, even among patients from the same family [16] or with the same molecular defect [17]. Furthermore, a marked change of phenotype with age from newborn period, infancy, childhood and adolescence to adulthood was classically documented, resulting in a mild phenotype in adult patients [3]. All these facts can contribute to misdiagnosis, especially in patients without congenital heart disease, with mild forms and/or at an older age.
In 1981, Duncan et al. [4] proposed a scoring system for NS diagnosis with 26 items devised on the basis of frequency and severity of NS features in 23 typical patients. This complex first scoring system was difficult for routine use by non-geneticist specialists. In 1993, Sharland et al. [18] proposed that the diagnosis should be based on the presence of typical facial features in a patient with normal chromosomes plus short stature (height <10th centile for sex and age) and/or cardiac defect and/or undescended testicles in males. These new criteria did not allow the diagnosis in patients with mild facial features. Finally, in 1994, van der Burgt et al. [16] proposed a simple and accurate scoring system for the diagnosis of NS based on the variable clinical NS presentation in one family (table 2). The van der Burgt scoring system is the most used model to select patients for molecular studies in recent studies [19,20,21]. In this scoring system, patients are first classified according to facial features as having typical or suggestive NS characteristics. Typical face and any other major sign or two minor signs establishes the diagnosis of NS, whereas patients with suggestive NS face need two major or three minor criteria to confirm the NS diagnosis. Analyses of clinical features in NS patients, who had their diagnosis confirmed by molecular study, demonstrated that no isolated clinical characteristic can ensure the diagnosis of NS; however, the van der Burgt et al. criteria, which take in account the facial features, growth pattern, chest deformity and cardiac defects, have been shown to be an accurate tool for NS diagnosis [22]. For these reasons, we recommend the use of this scoring system by endocrinologists during the evaluation of short stature children to facilitate the recognition of patients with NS.
![Noonan syndrome diagnostic criteria (adapted from van der Burgt et al. [16])](https://karger.silverchair-cdn.com/karger/content_public/journal/hrp/71/4/10.1159_000201106/2/m_000201106_t02.gif?Expires=1716587543&Signature=mXhj83C6bu-pXAm6E51u70ckAlDOgMrTHG3lCYUcOq~Zm5kXER5o8L~hprQyNyboIRJz9~wb2YTCNUsDtP7DuhHIIFrlzyhulu85lJn~Oxva7~0v7opkB4QGWs-dIqLNKPzBnwErkJkuqllH4K4nlLVnoMPaAU7DOcPr3sZ9eEn96hRB8pjmfbkneQlmjPr7lcZkiia8qLCl2s8SGVe2kv7lnSup7HWsbMRVr85c1wDiqhEZZWZiVExO6z7MzJBC2TBbjcsejc2vmt3ALkTFg1feg~9rK0KZHaim-8s9X0hmdvGixdosDO~BiOLV3LJKXfWq0eAoeViZ2j1jvziRiQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Genetic Heterogeneity of Noonan Syndrome
In 1994, Jamieson et al. [23] performed a genome-wide scan in a large Dutch NS family and demonstrated a linkage with several markers located at chromosome 12q22-qter. In parallel, they demonstrated the existence of genetic heterogeneity in NS by haplotype analysis of another NS family. Subsequent studies reduced the chromosome 12 interval which contains the NS candidate gene region [24, 25] and several positional candidate approaches were taken to identify the NS disease gene.
Finally, in 2001, Tartaglia et al. [26] demonstrated the presence of heterozygous missense mutations in PTPN11 (protein tyrosine phosphatase, non-receptor type 11; OMIM 176876), a gene mapped to chromosome 12q24.1, in patients with NS. Several further studies confirmed PTPN11 as the most affected gene in NS patients and demonstrated a mutation frequency of 38–100 and 37–52% in individuals with familial and sporadic NS, respectively [7, 19, 27, 28].
Germline PTPN11 mutations have also been detected in Leopard syndrome (OMIM 151100) [12, 13], Noonan-like/multiple giant cell lesion syndrome (OMIM 163955) [29] and in patients with isolated congenital heart disease [30] showing that mutations in this gene exhibit a broad phenotype spectrum. Furthermore, it is noteworthy that, during the screening of mutations in parents and relatives of NS patients with PTPN11 mutations, some individuals carried the same PTPN11 mutations found in the index case, but exhibited mild NS phenotypes that did not fulfill the NS diagnostic criteria [17, 26, 31, 32]. For these reasons, we investigated 50 children with idiopathic short stature that presented some NS-associated signs, without fulfilling the van der Burgt et al. criteria for NS diagnosis. No mutations were found in this cohort, suggesting that PTPN11 mutations are not involved in the pathogenesis of idiopathic short stature [22].
Specific somatic PTPN11 mutations were also found in leukemia patients, especially in juvenile myelomonocytic leukemia [33]. Additionally, somatic mutations are identified at low frequency in several human cancers [34].
The PTPN11 protein product, SHP-2 (Src homology region 2-domain phosphatase 2), is a ubiquitously expressed cytoplasmic protein member of a subfamily of protein tyrosine phosphatases that contains two Src homology 2 (C-SH2 and N-SH2) domains. Through SH2 domains, SHP-2 binds to activated receptors and adapter proteins that present specific phosphorylated tyrosines. In addition to tyrosine phosphatase actions, SHP-2 may also act as an adapter molecule through phosphorylation of a tyrosine residue at the amino terminus region, thus working as a docking site for other SH-2-containing molecules [35]. Both functions, tyrosine phosphatases and adapter molecules, are obviously relevant to several intracellular signal pathways, including signal transduction of growth factors and cytokines [36, 37]. From the endocrinological point of view, SHP-2 is implicated in GH [38, 39], IGF-1 [40], insulin [41] and leptin [42] signaling. The effects of NS PTPN11 mutation on these hormone actions are still to be evaluated.
The PTPN11 mutations identified in patients with NS or leukemia are predicted to be gain-of-function changes that augment the capacity of tyrosine dephosphorylation [26]. The SHP-2 mutants positively regulate the signal flux through the RAS/MAPK pathway induced by EGF (epidermal growth factor), FGF (fibroblast growth factor), IL-1 (interleukin-1) and TNF-1 (tumor necrosis factor-1), but negatively regulate JAK/STAT signaling [7]. Interestingly, somatic PTPN11 mutations associated with leukemia presented a higher phosphatase activity than germline mutations identified in NS patients [20, 36]. Additionally, somatic mutations commonly found in leukemias rarely occur as germline mutation in NS patients and vice versa [20, 36], suggesting that somatic PTPN11 mutations with higher tyrosine phosphatase activity and consequently with more leukemogenic properties could cause embryonic lethality if they occur as germline mutations.
Several other molecules in the RAS/MAPK pathway were involved in NS patients without PTPN11 mutations: germline KRAS [43], SOS1 [44, 45], RAF1 [46, 47] and MEK [15] mutations were found in 2.3, 21, 10 and 4.3% of NS patients who did not harbor mutations in PTPN11 gene, respectively. Furthermore, mutations in other RAS/MAPK molecules were also found in patients with NS-like syndromes (table 3). All these mutations, except those found in patients with Leopard syndrome [12, 13], are characterized by an increase of constitutive function of each mutated protein and consequently increased signal transduction via RAS/MAPK pathway. Due to the clinical and molecular disease mechanisms and similarities between NS and NS-like disorders, it is possible to categorize these syndromes as a group of disorders caused by RAS/MAPK pathway dysregulation.

RAS-MAPK Pathway
RAS proteins (HRAS, NRAS and KRAS) are small guanosine-binding proteins which act as signal switch molecules that integrate extracellular inputs and activate downstream effectors (fig. 3) [reviewed in [8, 48]]. Cell stimulation promotes cycling between inactive GDP-bound to active GTP-bound conformations (RAS-GDP and RAS-GTP). The counterbalancing activities of guanosine nucleotide exchange factors (GNEFs), which facilitate RAS-GTP conformation, and GTPase-activating proteins (GAPs), which increase the RAS intrinsic GTPase activity and act in favor of the RAS-GDP conformation, control RAS activity in vivo.
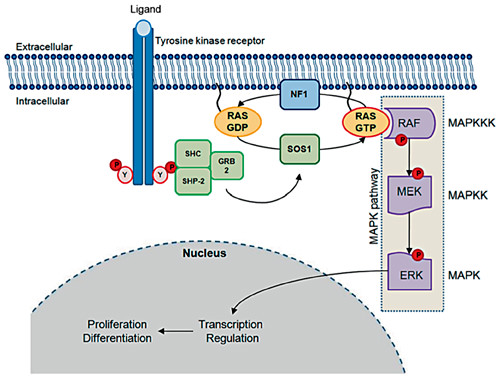
RAS/MAPK pathway. Tyrosine-phosphorylated domains of activated tyrosine kinase receptors act as docking sites for several intracellular proteins, which contain SH2 domains such as SHC, SHP-2 and GRB2. These molecules recruit SOS1 which promotes cycling between inactive RAS-GDP to active RAS-GTP. RAS-GTP directly activates the MAPK pathway (RAF-MEK-ERK cascade). ERK kinase can phosphorylate both cytosolic and nuclear substrates, which include transcription factors that control the cell cycle. SHC = Signaling and transforming protein containing Src homology 2 and 3 (SH2 and SH3) domains; SHP-2 = Src homology region 2-domain phosphatase 2; GRB2 = growth factor receptor-bound protein 2; SOS 1 = son of sevenless 1; NF1 = neurofibromin; RAS = rat sarcoma viral oncogene homolog; RAF = murine sarcoma viral oncogene homolog; MEK = mitogen-activated kinase kinase; ERK = mitogen-activated kinase.
RAS/MAPK pathway. Tyrosine-phosphorylated domains of activated tyrosine kinase receptors act as docking sites for several intracellular proteins, which contain SH2 domains such as SHC, SHP-2 and GRB2. These molecules recruit SOS1 which promotes cycling between inactive RAS-GDP to active RAS-GTP. RAS-GTP directly activates the MAPK pathway (RAF-MEK-ERK cascade). ERK kinase can phosphorylate both cytosolic and nuclear substrates, which include transcription factors that control the cell cycle. SHC = Signaling and transforming protein containing Src homology 2 and 3 (SH2 and SH3) domains; SHP-2 = Src homology region 2-domain phosphatase 2; GRB2 = growth factor receptor-bound protein 2; SOS 1 = son of sevenless 1; NF1 = neurofibromin; RAS = rat sarcoma viral oncogene homolog; RAF = murine sarcoma viral oncogene homolog; MEK = mitogen-activated kinase kinase; ERK = mitogen-activated kinase.
SOS1 (son of sevenless, drosophila, homolog 1 – OMIM 182530), the major GNEF, is recruited to protein complexes that assemble on activated growth receptors. SOS1 binds to either RAS-GTP or RAS-GDP and displaces guanine nucleotide. Because GTP is much more abundant than GDP in the cytosol, this nucleotide exchange increases intracellular RAS-GTP levels. In its GTP-bound form, RAS can activate several intracellular pathways, including the MAPK pathway. The RAF-MEK-ERK cascade is the best characterized RAS effector pathway. There are three RAF serine/threonine kinases (ARAF, BRAF and RAF1) that activate the MEK-ERK kinase cascade. ERK kinase can phosphorylate both cytosolic and nuclear substrates, which include transcription factors that control the cell cycle [8, 48].
Mutations in several molecules involved in this cascade were identified in NS and other related syndromes (table 3). It has been proposed that these disorders should be classified together as neuro-cardio-facial-cutaneous syndromes based on a constellation of similar phenotypic features and the central role of hyperactive RAS/MAPK in their pathogenesis [49]. Based on genotype-phenotype correlation, three clusters of genes were proposed [15]:
The first group comprises genes outside the RAS-RAF-MEK chain, which encompasses those upstream of RAS. Most patients with PTPN11 or SOS1 mutation have NS or Leopard syndrome. NF1 is caused by mutations in neurofibromin, a GAP protein involved in RAS inactivation. Mutations in this group of genes usually lead to a NS phenotype, with a low rate of mental impairment and a low rate of keratinization disorders, but a tendency to patchy skin hyperpigmentation, and, at least for NF1 and PTPN11, a slightly increased risk of leukemia [15].
The second group comprises KRAS and the cascading genes downstream. Mutations in these genes usually affect the cognitive functions, have more influence on somatic growth, skin redundancy, keratinization, and hair development, and usually result in a CFC syndrome phenotype. Malignancy risk appears to be low, but could include leukemias [15].
The third group is restricted to HRAS. Diffuse hyperpigmentation, papillomata, chaotic atrial fibrillation and a tendency for soft-tissue tumors are the most distinguishing phenotypic features in this group and usually result in a Costello syndrome phenotype [15].
Germline Mutations in Genes of RAS/MAPK Pathway and Risk of Neoplasia
Somatic activating RAS mutations occur in ∼30% of human cancers: mutations in KRAS are common in pancreatic, colorectal, endometrial, biliary tract, lung and cervical cancer, KRAS and NRAS mutations are prevalent in myeloid malignancies, whereas NRAS and HRAS mutations are found in melanoma and bladder cancer, respectively [8]. BRAF mutations are also frequently found in thyroid, colorectal and ovarian cancers [8] and somatic PTPN11 mutation is responsible for one third of juvenile myelomonocytic leukemia (JMML) and is less frequent in other leukemias or solid tumors [34].
In contrast with higher malignancy potential of somatic mutations in RAS/MAPK components, germline mutations presented more variable effects and depend on which specific genes are affected. Patients with Costello syndrome are predisposed to rhabdomyosarcoma, ganglioneuroblastoma and bladder cancer due to germline HRAS mutations, whereas patients with NF1 mutations frequently presented neurofibromas and show an increased risk of neurofibrosarcoma, astrocytoma, pheochromocytoma and JMML [8].
NS patients principally present an increase in the prevalence of JMML and multiple giant cell lesions (MGCL), although both conditions affect only a small percentage of patients. It is postulated that PTPN11 mutations associated with NS-JMML present a higher increase in tyrosine phosphatases activity than mutations only associated with NS and not found in leukemias [20, 36, 50]. MGCL are benign tumor-like lesions most frequently affecting the jaws and are associated with PTPN11, SOS1, BRAF and MEK1 mutations [51]. It is postulated that dysregulation of the RAS/MAPK pathway represents the common and basic molecular event which predisposes to MGCL.
The finding that the same molecular defects can be found in NS patients with or without concomitant JMML or MGCL indicates that NS-related heterozygous mutations in RAS/MAPK pathway components are not sufficient to produce both conditions and an additional unknown factor (modifier gene, epigenetic factors or somatic second hit) is necessary for JMML or MGCL development [51].
RAS/MAPK Pathway Dysregulation and Its Consequences for the Endocrine System
The consequences of NS and NS-related disorders with molecular RAS/MAPK pathway defects for the endocrine system remain mostly unexplored. One of the cardinal signs of NS is short stature [3, 4], although the physiopathological cause of growth impairment remains unclear. GH is essential for normal post-natal growth and exerts its action after binding to a specific receptor that phosphorylates several tyrosine residues located in the intracellular domain. Tyrosine dephosphorylation leads to the physiological interruption of the GH pathway. It has been consistently documented that SHP-2, the PTPN11 gene-encoded protein, negatively regulates GHR-JAK2-STAT5 signaling [39, 52]. Thus, the increased tyrosine phosphatase action of the SHP-2 protein, observed in PTPN11-mutated NS children, is expected to cause decreased GH action and consequently negatively influence individual stature. We demonstrated that NS children with PTPN11 gain-of-function mutations presented lower growth velocity and lesser height SDS gain during GH therapy than patients with NS without PTPN11 mutations [10]. These clinical findings were also observed in two other studies [9,10,11], suggesting that PTPN11 mutations can cause partial GH insensitivity at post-receptor level and appears to be a pharmacogenetic predictor of GH responsiveness [53].
Additionally, NS patients with SOS1 mutations, a molecule that was not directly involved in the GHR-JAK2-STAT5 pathway, more frequently have normal height [44, 45]. These NS patients also presented a high frequency of fetal macrosomia and macrocephaly, possibly due to the increase in the RAS-MAPK pathway caused by NS-associated SOS1 mutations and its positive influences on fetal growth [44, 45].
Patients with NS often present delayed puberty and NS boys frequently have cryptorchidism. Several cytokines, tyrosine and G-protein-coupled receptors are involved in the hypothalamic-pituitary-gonadal axis. Insulin acts through a tyrosine kinase receptor, and thus, the reproductive and metabolic effects of different NS-related molecular defects are still to be elucidated.
In summary, NS is a common condition and an important differential diagnosis in children with short stature, cryptorchidism and delayed puberty, conditions frequently seen by endocrinologists. Germline mutations that affect several components of RAS-MAPK pathway are involved in the pathogenesis of NS and can be diagnosed by molecular studies. Genotype-phenotype correlation studies are still necessary to better characterize the effect of RAS/MAPK pathway dysregulation on endocrine and metabolic systems.
Acknowledgments
This work was supported by grants from Fundacao de Amparo a Pesquisa do Estado de São Paulo (FAPESP) (05/04726-0 and 07/59555-0 to A.C.M.) and from Conselho Nacional de Desenvolvimento Cientifico e Tecnologico (CNPq) (301246/95-5 to B.B.M., 300938/06-3 to I.J.P.A. and 307951/06-5 to A.A.L.J.). The authors thank Prof. Martin O. Savage for his useful suggestions.
