

Abstract
It has been reported that patients with spinocerebellar degenerations (SCDs) have cognitive dysfunction as well as limb and truncal ataxia, dysarthria and dysphagia. We review cognitive dysfunction in common types of SCD, including spinocerebellar ataxia types 1, 2, 3, 6, and 17, dentatorubral-pallidoluysian atrophy, Friedreich’s ataxia, and multiple system atrophy. There are few studies that address cognitive function in SCD. Although there are few comparison studies among the various SCDs, cognitive dysfunction may be more common and severe in spinocerebellar ataxia type 17 and dentatorubral-pallidoluysian atrophy. While cognitive dysfunction in SCD appears to represent frontal dysfunction, the mechanisms of cognitive dysfunction have not been directly clarified. Nevertheless, various lesions, including those in the cerebrocerebellar circuitry, cortico-striatal-thalamocortical circuitry, and the frontal lobe, may influence cognitive function to various degrees for each disease.
Introduction
Spinocerebellar degenerations are neurodegenerative diseases that involve the cerebellum, brain stem, spinal cord, and basal ganglia to various degrees. Patients with spinocerebellar degeneration display limb and truncal ataxia, dysarthria, dysphagia, extrapyramidal sign (dystonia, rigidity, and bradykinesia), pyramidal sign, and autonomic disorder. Although they are also reported to have cognitive impairment, this remains controversial.
Spinocerebellar degeneration includes both sporadic and hereditary forms. Most cases of sporadic spinocerebellar degeneration are now considered to be multiple system atrophy (MSA). Although some of these patients have symptoms such as cerebellar cortical atrophy, they may subsequently display extrapyramidal signs and autonomic disorder similar to MSA [1]. Hereditary spinocerebellar degeneration consists of autosomal dominant spinocerebellar degeneration, including spinocerebellar ataxia (SCA) types 1, 2, 3, and 6 and autosomal recessive spinocerebellar degeneration, such as Friedreich’s ataxia (FA) [2, 3]. Recently, genes responsible for these diseases were cloned [4,5,6,7]. Although cognitive function in SCA patients were examined before genetic analyses became available, these studies were controversial as they included many types of SCA.
On the other hand, there have been several recent clinical reports of patients with cerebellar lesions and cognitive dysfunction [8], and the importance of the cerebellum in cognitive function has been recognized. Here, we review cognitive dysfunction in common types of spinocerebellar degenerations, including SCA1, SCA2, SCA3, SCA6, dentatorubral-pallidoluysian atrophy (DRPLA), SCA17, FA, and MSA.
Spinocerebellar Ataxia Type 1
SCA1 is characterized clinically by cerebellar ataxia, dysarthria, and bulbar dysfunction, with variable degrees of brain stem oculomotor signs, ophthalmoplegia, hyperreflexia, spasticity, extensor plantar responses, peripheral neuropathy and fasciculations.
Although there are some studies that examined cognitive function in SCA1 patients, most of them were reported before genetic analysis became available. In family members of a large SCA1 kindred, Kish et al. [9] found decreased Mini-Mental State Examination and verbal IQ scores, suggesting general intellectual impairment. They also revealed impairment of memory, naming, visuospatial function and executive function. Tang et al. [10] found that clinically, dementia was less frequent in SCA1 than in the other SCAs. The first comprehensive study of cognitive function in genetically confirmed SCA1 was described by Bürk et al. [11]. They examined 14 patients with SCA1 using a neuropsychological test battery. Verbal memory and executive function were significantly impaired in these patients. In the verbal memory test, SCA1 patients reproduced significantly fewer items than did controls during the immediate recall of the consecutive categories, randomized categories and uncategorized lists and during the delayed recall of the consecutive categories and uncategorized lists. Test performance was not related to either trinucleotide repeat length or disease duration. A comparison of cognitive dysfunction among SCA1, SCA2 and SCA3 patients revealed that executive dysfunction was more prominent in SCA1 patients compared with other SCA types [12]. It was also reported that depressive and memory symptoms were found in 25 and 42% of SCA1 patients, respectively [13].
The mechanism of cognitive impairment in SCA1 remains unclear. There are only a few studies that address supratentorial change in SCA1. Neuropathological findings were reported to be loss of Purkinje cells and variable loss of granule cells in the cerebellar cortex, neuron loss and moderate gliosis in the dentate nuclei, neuron loss and severe gliosis in the inferior olives and the pontine nuclei, loss of neurons and pigment deposition in the substantia nigra, and variable involvement of the putamen, pallidum, and subthalamic nucleus, while the cerebral cortex was normal [3, 14]. Gilman et al. [15] showed that SCA1 patients exhibited hypoperfusion of the cerebral cortex, caudate nucleus, putamen and thalamus, as well as cerebellum and brain stem. Cognitive impairment in SCA1 is therefore contingent upon damage to different parts of this cerebrocerebellar circuitry. In addition to cerebellar connections, cognitive impairment in SCA1 could result from disruption of the basal ganglia-thalamocortical circuitry.
Spinocerebellar Ataxia Type 2
Ataxia is the predominant clinical manifestation in SCA2, although the SCA2 phenotype differed from those of other SCAs with higher frequencies of slowed ocular movements, postural and action tremor, myoclonus, and hyporeflexia. Several clinical investigations disclosed the frequency of dementia in SCA2 patients to be between 19 and 42% [10,16,17,18], although in a comparison of various SCAs, Bürk et al. [12] did not find significantly lower scores on neuropsychological tests in SCA2 patients. It has been reported that SCA2 patients have various cognitive disorders, such as developmental delay [19] and olfactory impairment [20]. In another study, Bürk et al. [21] systematically addressed the issue of cognitive function in genetically confirmed SCA2 by means of comprehensive neuropsychological testing. Twenty-five percent of the SCA2 subjects showed evidence of dementia. Even in nondemented SCA2 subjects, there was evidence of verbal memory and executive dysfunction. Demented SCA2 patients exhibited significant impairment of the immediate recall of uncategorized and randomized category lists and the delayed recall of consecutive category list type. There was no relationship between test performance and trinucleotide repeat length. Le Pira et al. [22] partially confirmed these results, but they also found defects in attention as well as nonverbal intelligence task. Fernandez-Ruiz et al. [23] tested 43 SCA2 patients and their matched controls in prism adaptation, a kind of visuomotor learning task. SCA2 patients showed an impaired strategic control that affected the adaptation rate, but a normal spatial realignment measured through the aftereffect.
Although the mechanism of cognitive impairment in SCA2 remains unclear, disruption of the dopaminergic nervous system was reported in several studies of SCA2 patients. A reduction in striatal dopamine transporters resembling that observed in patients with Parkinson’s disease (PD) was found in SCA2 patients, using SPECT [24,25,26] and PET [27]. Another PET study in SCA2 patients showed reduced fluorodopa uptake in the striatum and normal dopamine D2/D3 receptor density, all typical of idiopathic PD [28]. In an [18F]-fluorodeoxyglucose PET study, decreased regional cerebral glucose metabolism was found not only in the cerebellum but also in the brain stem and the parietal cortex of patients with SCA2 [27]. Using voxel-based morphometry, Brenneis et al. [29] observed significant volume loss not only in infratentorial regions, such as the cerebellar hemispheres, vermis, pons, mesencephalon and thalamus, but also in several supratentorial areas, such as the right orbitofrontal cortex, right temporomesial cortex and the primary sensorimotor cortex bilaterally in SCA2 patients. This involvement of widespread multiple regions was confirmed by neuropathological examination of SCA2 brains, which revealed degenerations mainly in the cerebellar vermis and hemispheres, including Purkinje cells and granular cells, middle cerebellar peduncles, inferior cerebellar peduncle, substantia nigra, inferior olives, pontine nuclei, Clark’s nuclei, and dorsal columns. Neuropathological progression later included neuronal loss even in the neocortex [3, 16, 30]. These degenerations might result in the more common morbidity of dementia.
Spinocerebellar Ataxia Type 3 (Machado-Joseph Disease)
The wide range of clinical manifestations in SCA3 include cerebellar ataxia, hyperreflexia, spasticity, eyelid retraction, ophthalmoplegia, parkinsonism, dystonia, hyporeflexia, fasciculations, myotrophy, sensation loss, restless legs syndrome, cold intolerance and nocturia. Several clinical investigations showed that dementia in SCA3 might be less frequent (5–13%) than in other SCAs [10, 18, 31]. However, it has been reported that SCA3 patients do have various cognitive and psychiatric disorders, such as REM sleep behavior disorder [32] and delirium [33]. Maruff et al. [34] examined cognitive function in 6 patients with genetically confirmed SCA3 using a series of subtests from the Cambridge Neuropsychological Test Automated Battery. These patients had deficits in visual attentional function that were characterized by a slowing of the processing of visual information when task demands were high and an inability to shift attention to previously irrelevant stimulus dimensions to discriminate between complex stimuli. Zawacki et al. [35] gave 6 individuals with SCA3 a battery of neuropsychological tests, and found relative impairments on timed verbal attention, verbal fluency and set-shifting. Kawai et al. [36] examined 16 genetically confirmed SCA3 patients using neuropsychological tests, and found more extensive cognitive impairments, including those of verbal and visual memory, visuospatial and constructional abilities and verbal fluency. None of these impairments correlated with CAG repeat length or disease duration. It was also reported that depressive symptoms and apathy were common in SCA3 patients [13,35,36,37]. Recently, it was revealed that SCA3 patients were impaired on a Theory of Mind task, although they were not impaired on tasks requiring attribution of emotions or judgments of behavior in social situations [38].
The neuropathological findings of SCA3 consist of degeneration in the dentate nucleus, superior cerebellar peduncle, globus pallidus, substantia nigra, subthalamic nucleus, red nucleus, pontine nuclei, oculomotor nucleus, medial longitudinal fasciculus, anterior horn, spinocerebellar tracts, Clark’s nuclei, intermediolateral column, and lateral reticular nucleus [3, 37, 39]. However, it was found that the nuclear accumulations of expanded polyglutamine stretches also involved many neurons covering a wide range of central and peripheral nervous system regions, including the cerebral cortex and thalamus, that have been categorized previously as spared regions by conventional pathological studies [40]. These lesions, newly recognized by polyglutamine immunohistochemistry, may be responsible for the cerebral cortical dysfunctions in SCA3 patients. Consistent with the results of this pathological study, the regional cerebral glucose metabolism or perfusion in SCA3, studied with PET or SPECT, was found to be significantly decreased in the entire cerebral cortex [41], occipital cortex [42], frontal, temporal and parietal lobes [43] aswell as in the cerebellar hemispheres, vermis, and brain stem. Murata et al. [44] found similar results using MRI; not only were the widths of the superior cerebellar peduncles reduced and the transverse diameters of the globus pallidus and pons diminished, but the frontal and temporal lobes were also atrophied. As in SCA2, disruption of the dopaminergic nervous system has also been reported in SCA3 patients. SPECT [45] and PET [27] revealed significantly decreased dopamine transporter binding in both the putamen and the caudate nucleus in most SCA3 patients. Also, fluorodopa uptake in striatum was reduced, but dopamine D2/D3 receptor density was normal in most SCA3 patients [41, 46]. These results may indicate that SCA3 patients exhibit dysfunction, not only in the regions with apparent pathological involvement such as cerebellum, brain stem and nigrostriatal dopaminergic system, but also in the cerebral cortex and the striatum. The basis for cognitive dysfunction in SCA3 may be multifactorial, including cerebral cortical dysfunctions due to the nuclear inclusions, disruption of cerebrocerebellar circuitry, and cortico-striatal-thalamocortical circuitry.
Spinocerebellar Ataxia Type 6
The clinical features in these patients consist of cerebellar ataxia of the limbs, trunk and gait, horizontal nystagmus, and dysarthria without extrapyramidal signs, ophthalmoplegia, and peripheral neuropathy. There have been very few studies addressing cognitive function in SCA6 patients because researchers assume, and it is indeed true, that SCA6 patients are rarely impaired in cognitive function (0–20%) [18,47,48,49]. Globas et al. [50] examined cognitive function in 12 patients with genetically confirmed SCA6, but found only mild deficits in fronto-executive tasks that were not significantly different from those in controls. However, Suenaga et al. [51] examined cognitive function in 18 patients with genetically confirmed SCA 6, and showed that verbal fluency and immediate visual memory were markedly impaired. These cognitive dysfunctions did not correlate with CAG repeat length. Moreover, SCA6 patients were impaired on a Theory of Mind task [38].
Neuropathologically, SCA6 is characterized by almost exclusive cerebellar involvement, particularly selective loss of the cerebellar Purkinje cells and mild to moderate loss of granular cells and neurons in the dentate nucleus, while cortical structures and basal ganglia are spared [3,52,53,54]. Thus, SCA6 might represent an excellent model for investigating the cerebellar contribution to cognition. However, imaging of regional cerebral glucose metabolism and cerebral brain perfusion in SCA6 patients has also produced conflicting results. Soong et al. [55] found that glucose metabolism rates were significantly lower not only in the cerebellar hemispheres, but also in the brain stem, basal ganglia, and frontal, temporal, and occipital cerebral cortices, while Honjo et al. [56] found that regional cerebral blood flow was decreased only in the cerebellar vermis and hemispheres in SCA6 patients. Recently, statistical parametric mapping demonstrated that brain metabolism and perfusion were diminished in the cerebellum and the prefrontal cortices in SCA6 [57, 58]. Kawai et al. [57] revealed that neuropsychological test scores in SCA6 patients were significantly correlated with a decrease in prefrontal perfusion, and suggested that the cognitive dysfunctions in SCA6 patients may result from prefrontal dysfunction, based on the functional deactivation of the cerebello-ponto-thalamo-cerebral pathways.
Dentatorubral-Pallidoluysian Atrophy
DRPLA is a spinocerebellar degeneration that is relatively prevalent in Japan. It is characterized by various combinations of ataxia, choreoathetosis, myoclonus, epilepsy and dementia that resemble symptoms of Huntington’s disease [3]. Patients commonly show psychiatric symptoms, such as instability in mood, irritability, apathy, childish behavior, and euphoria, and less frequently, delusion and visual or auditory hallucinations. Cognitive profiles in DRPLA are generally compatible with those frequently seen in patients with subcortical dementia, characterized by relatively mild memory deficit, psychomotor retardation, and abnormal executive function. MRI or CT scans often reveal atrophy of the cerebellum and brain stem, microcalcification of the basal ganglia, and leukodystrophic changes, although they are variable. Ikeuchi et al. [59] reported frequent observations of ataxia and dementia, and significant correlations between the size of CAG repeats, and dementia and psychiatric symptoms. Schöls et al. [3] recommended that genetic analyses for DRPLA be directed to patients with dementia or psychosis as well as those with epilepsy, myoclonus or chorea in autosomal dominant cerebellar ataxias. Although we found no comprehensive neuropsychological examination studies, we assumed that the profile was similar to those of other SCAs.
Neuropathological examination revealed combined degeneration of the dentatorubral and pallidoluysian systems [60], and degeneration of the fastigio-vestibular system [3], while loss of neurons in cerebral cortex was mild. In some cases, dramatic myelin loss in the cerebral white matter was accompanied by axonal preservation and mild atherosclerotic changes in vascular blood vessels [61]. These results were consistent with those of neuroradiological studies [62].
Quantitative evaluations of DRPLA cases revealed no significant loss of neurons in the nucleus basalis of Meynert, and no clinicopathological correlation between dementia and involvement of the nucleus basalis of Meynert [63]. Yamada et al. [64] reported increased nuclear labeling in large populations of neurons in many CNS regions, including cerebral cortex, globus pallidus, subthalamic nucleus, oculomotor nucleus, red nucleus, substantia nigra, pontine nuclei, vestibular nucleus, cerebellar dentate nucleus, and spinal anterior horn. The results suggest that the novel lesion distribution revealed by the diffuse nuclear labeling may be responsible for a variety of clinical features, including dementia, in DRPLA [64].
Spinocerebellar Ataxia Type 17
SCA17 is a newly described, dominantly inherited disease that is caused by a CAG or CAA repeat expansion in the TATA-binding protein, a transcription factor [65]. The presentation is variable, but most individuals present between the ages of 20 and 30 with ataxia and dementia [3]. MRI or CT findings indicated diffuse cortical and cerebellar atrophy in all patients examined [65]. The clinical features of SCA17 patients, manifested by dementia and psychiatric abnormalities including depression, personality changes, aggressiveness, negligence of personal hygiene, delusional thoughts, and hallucinations, differ from those of other SCA types [66, 67]. Although this genotype is rare: 15 SCA17 patients in 1,318 SCA patients [67], it is important, because genetic analyses for SCA17 should be directed also to the patients with dementia or psychosis, in addition to those with cerebellar ataxia [3]. The full phenotypic variability has not been determined [68, 69].
Conventional neuropathological examinations revealed shrinkage and moderate loss of small neurons with gliosis in the caudate nucleus and putamen. Similar but moderate changes were detected in the thalamus, frontal cortex and temporal cortex. Moderate Purkinje cell loss and an increase in Bergmann glia were seen in the cerebellum [65,66,67, 70]. Intranuclear neuronal inclusion staining with anti-TATA-binding protein and anti-polyglutamine were much more widely distributed throughout the brain gray matter than in other SCAs [67]. In neuroimaging studies, SCA17 patients showed significantly reduced glucose metabolism and dopamine transporters in the basal ganglia, suggesting disruption of the dopaminergic nervous system [71, 72]. Lasek et al. [73] reported degeneration in the bilateral cerebellar posterior lobe, bilateral putamen, left thalamus, left inferior frontal gyrus, right inferior parietal lobule and left cuneus, among others. They also showed that scores on the Mini-Mental State Examination were significantly correlated with atrophy of the left ventral striatum. This broad involvement of the CNS may result in the dementia and psychiatric symptoms seen in SCA17 patients.
Friedreich’s Ataxia
FA [for review, see ref. [2]] is the most common of the autosomal recessive ataxias, occurring with a frequency of approximately 1 in 30,000–50,000 in the Caucasian population. Most cases of FA are caused by loss of function mutations in the frataxin gene, located on chromosome 9q13. Neuroimaging does not show progressive cerebellar degeneration, unlike the autosomal dominant hereditary ataxias. It primarily affects the spinocerebellar tracts, posterior columns, and to a lesser extent the corticospinal tracts. Clinical manifestations include gait and limb ataxia, dysarthria, pes cavus, lower extremity weakness, cardiomyopathy, and diabetes [2].
There are few studies addressing the cognitive profiles of FA. Slowed information processing speed, visuospatial deficits, impaired verbal learning and executive dysfunctions have been described in recent studies [74]. Most of them were reported before genetic analysis became available. The first comprehensive study of cognitive function in genetically confirmed FA was conducted by Mantovan et al. [75]. Neuropsychological and affective changes in 13 FA patients included lower IQ scores than controls, and impairments of visuoconstructive abilities, verbal fluency, attention, information speed processing and planning, and implicit learning. They also described a personality pattern consisting of increased irritability, poor impulsive control and blunted affect. The GAA expansion size did not correlate with neuropsychological performance. De Nobrega et al. [76] described differential impairments in semantic, phonemic, and action fluency performance in FA. There was no correlation between the degree of atrophy and either neuropsychological assessment or the score on clinical scales. Degeneration in FA first occurs in the spinal cord. Cerebellar degeneration is usually mild, and cerebral atrophy occurs in late stages of FA [77]. By examining the correlation between cognitive dysfunctions, and brain atrophy, perfusion and metabolism, the mechanism of cognitive dysfunction in FA may be revealed.
Multiple System Atrophy
MSA is a sporadic neurodegenerative disease that presents with parkinsonism, cerebellar ataxia, autonomic failure, and pyramidal signs of varying severity during the course of the illness [78,79,80]. Diagnostic criteria for MSA proposed by a Consensus Conference in 1998 [81] recommend designating patients as having MSA-P if parkinsonian features are predominate or MSA-C if cerebellar features predominate.
Although dementia consistent with the Diagnostic and Statistical Manual of Mental Disorders (DSM IV) is an exclusion criterion for the diagnosis of MSA [81], some studies have reported that MSA patients have a cognitive decline when compared with controls [82,83,84,85,86,87,88]. MSA-P is the more common manifestation of MSA in Western populations [89, 90]; therefore, cognitive dysfunction in MSA-P had attracted considerable attention, particularly in comparison with PD and/or progressive supranuclear palsy [82,83,84,86,87,88]. MSA-P patients showed significant deficits in the tests previously shown to be sensitive to frontal lobe dysfunction [83, 85, 86, 88]. There are several studies demonstrating that MSA-P patients have apraxias [91,92,93] or depressive state [94,95,96]. In comparisons among PD and MSA-P and progressive supranuclear palsy, although results were not consistent, all studies showed patients with progressive supranuclear palsy were most severely impaired in cognitive function. This may be due to which characteristic MSA and PD patients were matched in their studies (severity of motor symptoms, age or disease duration). Dujardin et al. [82] reported that MSA patients have more severe and diffuse impairments than both severity of motor symptom-matched and disease duration-matched PD patients. Using discriminant function analysis, Lange et al. [97] found that verbal fluency tasks discriminated among the three patient groups at a level significantly exceeding chance.
Recently, Bürk et al. [98] examined comprehensive neuropsychological tests in MSA-C patients and found impaired verbal memory and verbal fluency. Kawai et al. [99] compared cognitive profiles of MSA-C and MSA-P patients. Although they had similar profiles, on the whole, MSA-P patients were more severely impaired in cognitive functions than MSA-C patients.
In recent years, some cases of MSA were reported to have cerebral cortical atrophy with white matter involvement [100,101,102,103,104]. The main pathological change comprised cell loss and gliosis in the putamen, substantia nigra, locus ceruleus, inferior olives, pontine nuclei, cerebellar Purkinje cells, and intermediolateral cell columns of the spinal cord [105]. Sites mildly or rarely involved included cerebral cortex, thalamus, subthalamic nucleus, caudate nucleus, globus pallidus, dentate nucleus, nucleus ambiguous, vestibular nuclei, anterior horn cells, and pyramidal tracts. However, oligodendroglial cytoplasmic inclusion-rich structures occurred in primary motor and higher motor areas of cerebral cortex, ‘pyramidal’, ‘extrapyramidal’ and cortico-cerebellar systems [106]. In MSA-C patients, a study using voxel-based morphometry revealed that gray matter was reduced in the bilateral cerebellum [107], and another study revealed that atrophy was observed not only in the cerebellum, but also in frontotemporal areas of both hemispheres [108]. The pathological background of cognitive impairments in these patients might be the widespread cortical involvement.
Using 1H-MR spectroscopy in MSA, Watanabe et al. [109] found that the N-acetylaspartate-to-creatine ratio in cerebral white matter, as well as in the pontine base and putamen, tended to decline in long-standing cases. In voxel-based morphometry, white matter atrophy in MSA-C was observed in pons and mesencephalon as well as in frontotemporal areas of both hemispheres [110]. Hence, the cognitive dysfunction in MSA may be also influenced by white matter involvement.
Other possible explanations for the cognitive deficits in MSA-C are based on the disruption of cerebrocerebellar circuitry, or on the disruption of a cortico-striatal-thalamocortical circuitry that links the frontal cortex to the basal ganglia and thalamus. In support of the latter, MSA patients exhibited significantly decreased dopamine transporter binding in the putamen [110,111,112].
To clarify the mechanism of cognitive dysfunction in MSA, Van Laere et al. [113] examined the relationship between brain perfusion and clinical parameters in MSA-P. Perfusion in the bilateral posterior associative cortex and posterior cingulate was negatively correlated with cognitive function. Kawai et al. [99] examined the relationship of cognitive function to brain perfusion in MSA-C and MSA-P, and showed that there were positive correlations between the scores of neuropsychological tests and perfusion in the prefrontal cortices in MSA-P patients and perfusion in the prefrontal cortices and cerebellar hemisphere in MSA-C patients. These results suggest that the mechanisms of cognitive impairment in MSA-C and MSA-P may be slightly different. We suggest that the performance on cognitive tasks in MSA-C patients is influenced by various lesions, including those in the cerebrocerebellar circuitry, cortico-striatal-thalamocortical circuitry, and the frontal lobe.
The Mechanism of Cognitive Impairment in Spinocerebellar Degenerations
Cognitive impairment in spinocerebellar degeneration patients is variable, but similar to that for prefrontal dysfunction (table 1). Schmahmann and Sherman [8] suggested that the cerebellum participates in the organization of higher order function, and behavioral changes in patients with lesions involving the cerebellum were characterized by impairment of executive functions, difficulties with spatial cognition, personality change, and language deficits [8]. The distribution of patients’ scores differed significantly from the normal distribution, with the most marked deviation from normal evident in the categories of executive (for example, distribution of Z-scores for the patients: –2.7 ± 1.8 in the Controlled Oral Word Association Test and –0.89 ± 0.76 in the Trail Making Test part B) and visual spatial function (for example, distribution of Z-scores for the patients: –5.9 ± 3.2 in the Rey Complex Figure test and –1.2 ± 0.9 in Block Design). Attention and orientation, and language functions more closely approximated a normal distribution of scores. They proposed the name ‘cerebellar cognitive affective syndrome’ for such cases. The clinical observations in these patients are supported by functional neuroimaging studies demonstrating cerebellar activation in cognitive tasks. Cerebellar activation has been observed during tests of word generation [114], processing of words [115], working memory [116], verbal memory [117], motor sequence learning [118], cognitive processing [119], classical conditioning [120], attention [121], mental imagery [122], sensory discrimination [123], and visual discrimination [124]. Neuropathological changes in spinocerebellar degeneration are various and include those in the cerebrocerebellar circuitry, cortico-striatal-thalamocortical circuitry and the frontal lobe; however, patients with spinocerebellar degeneration all have involvement in cerebellum. Cognitive dysfunction such as prefrontal dysfunction, which can be seen in all spinocerebellar degenerations, may result from the involvement in cerebellum, and represent the ‘cerebellar cognitive affective syndrome’. The fact that patients with SCA6, which is characterized by degeneration that is greatly restricted to the cerebellum with cortical structures spared, have cognitive dysfunction similar to those with other spinocerebellar degeneration, suggests that cognitive dysfunction in spinocerebellar degeneration includes ‘cerebellar cognitive affective syndrome’.
Summary of the neuropsychological and neuropathological test results in spinocerebellar degenerations
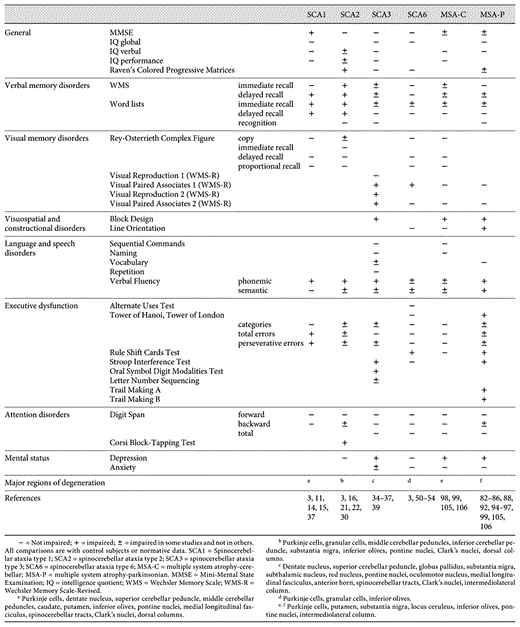
The mechanism of cognitive dysfunction in spinocerebellar degeneration has not been directly clarified. However, various lesions, including those in the cerebrocerebellar circuitry, cortico-striatal-thalamocortical circuitry, and the frontal lobe, influence cognitive function to various degrees for each disease. The pathology of spinocerebellar degeneration is various, and may be related to the diversity of cognitive dysfunction. Schmahmann and Sherman [8] found that cognitive and affective changes were clinically prominent in patients with lesions involving the posterior lobe of the cerebellum and that lesions of the anterior lobe of the cerebellum produced only minor changes in executive and visual-spatial functions [8]. Cognitive dysfunction in patients with spinocerebellar degeneration may be also influenced by the site of cerebellar degeneration. To address the background mechanism of cognitive dysfunction, Kawai et al. [57] examined the regions in which regional cerebral blood flow was significantly correlated with scores of neuropsychological tests on which SCA6 [57] and MSA [99, 113] patients showed impairment, after the influences of age, education, ataxia and parkinsonism were excluded. They found that there were correlations between the scores of neuropsychological tests and perfusion in the prefrontal cortices in patients with each diseases. These results suggest that cognitive impairments in spinocerebellar degenerations result from prefrontal dysfunction. However, disruption of the cerebrocerebellar circuitry or cortico-striatal-thalamocortical circuitry may also cause prefrontal dysfunction, so the mechanism of such dysfunctions remains unresolved. Thus, it is important to compare the various cognitive impairments with their specific neuropathologies.
Conclusion
We reviewed the cognitive dysfunctions in spinocerebellar degenerations, including SCA1, SCA2, SCA3, SCA6, DRPLA, SCA17, FA and MSA-C. Cognitive dysfunction in spinocerebellar degenerations was various, but appeared to represent frontal dysfunction. Cognitive dysfunction is not characteristic for each disease, but seems to have a similar profile. It is difficult to compare the cognitive dysfunctions of the spinocerebellar degenerations, because the progression of each disease is various. For example, SCA6 is relatively slowly progressive, while MSA-C is relatively rapidly progressive. Moreover, which characteristic is used for comparison matching (disease duration, age, or disease severity) also complicates the comparisons. However, it is considered that cognitive impairment is common in SCA17 and DRPLA, and rare in SCA6.
The mechanism of cognitive dysfunction in spinocerebellar degeneration has not been clarified [77]. The pathologies of spinocerebellar degeneration are diverse, and this diversity may be related to the natures of the cognitive dysfunctions. It is important to compare the cognitive impairments with their particular neuropathologies. Also, neuroimaging studies have revealed cortical atrophy, white matter involvement, brain hypoperfusion and hypometabolism in spinocerebellar degeneration; however, how the results are related to cognitive function is not clear. By evaluating the correlation between cognitive dysfunctions, and brain atrophy, perfusion and metabolism, the mechanism of cognitive dysfunction may be revealed.
