

Abstract
Medullary thyroid cancer (MTC) arises from neural-crest-derived parafollicular C cells of the thyroid gland and accounts for approximately 4% of all thyroid cancers. Up to 25–30% of MTC cases occur as inherited disorders while the remaining cases represent the sporadic form of the disease. In this review, the structure and signalling properties of the RET proto-oncogene in its wild-type and mutant forms, and its role in hereditary and sporadic MTC, are discussed. A full data search was performed through PubMed over the years 2000–2008 with the key words ‘medullary thyroid cancer, treatment, molecular biology, RET, molecular mechanism’, and all relevant publications have been included, together with selected publications prior to that date. We also review novel therapies for metastatic MTC, especially the tyrosine kinase inhibitors which have activity at multiple receptor subtypes, and summarize the current ongoing trials in this area. While such tyrosine kinase inhibitors, particularly those affecting RET activity such as vandetanib, sorafenib and sunitinib, are promising, the low rate of partial responses and absence of complete responses in all of the various trials of monotherapy emphasize the need for new and more effective single agents or combinations of therapeutic agents with acceptable toxicity.
Introduction
Medullary thyroid cancer (MTC) is a rare thyroid tumour first described in 1959 by Hazard et al. [1]. Medullary thyroid cancer arises from neural-crest-derived parafollicular C cells of the thyroid gland and accounts for approximately 4% of all thyroid cancers [2]. Up to 25–30% of MTC cases occur as inherited disorders while the remaining cases represent the sporadic form of the disease [3]. Hereditary MTC can present in three major subtypes: as part of multiple endocrine neoplasia type 2 (MEN 2A or MEN 2B), and as familial MTC (FMTC). MEN 2 is a genetic syndrome caused by germline mutations in the RETproto-oncogene and is transmitted in an autosomal dominant pattern. The most common form of hereditary MTC is MEN 2A (approximately 80–90% of patients with hereditary MTC) in which MTC occurs in 100%, while phaeochromocytoma and hyperparathyroidism are seen in approximately 50 and 20% of cases, respectively [4]. A minority of patients with MEN 2A develop characteristic cutaneous lichen amyloidosis or a limited form of Hirschsprung’s disease. MEN 2B comprises some 5–10% of MEN syndromes, with MTC seen in 100% and phaeochromocytoma in 50% of cases. Enteric ganglioneuromas, a marfanoid body habitus, distinctive mucosal neuromas of the tongue, lips and subconjunctival areas and medullated corneal-nerve fibres are also seen as features of MEN 2B. FMTC accounts for 5–15% of hereditary MTC cases. It is defined as the presence of MTC in kindreds with 4 or more affected members without involvement of adrenal and parathyroid glands [5]. Moreover, FMTC can occur as only C cell hyperplasia (CCH), without tumour formation [6]. Although rare, large families with both FMTC and MEN 2A have also been reported [7].
At presentation, hereditary MTC is usually bilateral and multicentric. Multifocal CCH is considered to be a precursor to invasive MTC in patients with hereditary disease [8]. In sporadic cases, the tumour is generally found as a single unilateral thyroid nodule or a palpable cervical lymph node. Finally, a minority of patients may present with systemic manifestations due to secretion of several peptides and substances, including calcitonin, chromogranin A, neurotensin, bombesin, somatostatin, vasoactive intestinal peptide, carcinoembryonic antigen (CEA) and calcitonin-gene-related peptide, and adrenocorticotropic hormone (ACTH) in the ‘ectopic ACTH syndrome’ [9].
The aggressiveness of MTC varies with the subtypes. While sporadic cases often present between 50 and 60 years, in a family with a confirmed diagnosis, a MEN 2A patient may present at the age of 5 years and upwards [10]. MEN 2B is the most aggressive hereditary form based on its development of MTC earlier in life, with cervical lymph node metastases being observed in patients as young as 3 years: it has a high mortality [11]. FMTC tends to have the least aggressive course among hereditary MTCs, often presenting between 20 and 40 years [12]. There are strong genotype-phenotype correlations in MEN 2 depending on the location of the RET mutation. Hence, classification of patients into different risk levels according to their RETmutation type and performing prophylactic thyroidectomy in these patients according to this classification before reaching a certain age have been recommended by different authors [13, 14]. However, these associations seem to occur particularly within a given family. Thus, some mutations correlating with aggressive MTC and leading to MTC-related deaths in some families were also reported in some FMTC families with no known cases of MTC-related deaths [3]. This suggests the importance of environmental and other genetic factors in the biological behaviour of MTC.
In this review, the structure and signalling properties of the RET proto-oncogene in its wild-type and mutant forms, and its role in hereditary and sporadic MTC, will be discussed. A full data search was performed through PubMed over the years 2000–2008 with the key words ‘medullary thyroid cancer, treatment, molecular biology, RET, molecular mechanism’, and all relevant publications have been included, together with selected publications prior to that date. Additionally, as most conventional therapies (chemotherapy, radiotherapy) are relatively ineffective for metastatic disease, novel therapeutic options for this challenging cancer type MTC will be reviewed and assessed.
Molecular Biology of MTC and the RET Proto-Oncogene
RET Biology and Signalling
The RET (REarranged during Transfection) gene was first identified in 1985 by Takahashi et al. [as mentioned in ref. [15]] as a proto-oncogene that can undergo activation by DNA rearrangement. The RET gene is located on chromosome 10q11.2 near the centromere and includes 21 exons [16]. It encodes a plasma-membrane-bound receptor tyrosine kinase that is mainly expressed in precursor cells of the neural crest and urogenital tract, with key roles in cell growth, differentiation and survival. RET is essential for the early development of the sympathetic, parasympathetic and enteric nervous systems, the kidney and spermatogenesis [17]. Inactivating germline RET mutations are found to be responsible for the development of Hirschsprung’s disease, a congenital absence of enteric neurons in the gastrointestinal tract. RETis classified as a proto-oncogene because an experimentally induced or a naturally occurring single activating RET mutation of one allele can lead to neoplastic transformation. Activating RET mutations or rearrangements are found in a number of cancers including MEN 2 and FMTC.
RET is a single-pass transmembrane receptor belonging to the tyrosine kinase superfamily. In the extracellular domain, there are 4 Ca2+-dependent cell adhesion (cadherin)-like domains and a juxtamembrane cysteine-rich region [18] (fig. 1). The intracellular region includes 2 tyrosine kinase subdomains (TK1 and TK2) that are involved in the activation of several intracellular signal transduction pathways. There are 3 different splicing variants of the carboxy-terminal tail of RET carrying 9 (RET9-short isoform), 43 (RET43-middle isoform) and 51 (RET51-long isoform) distinct amino acids. RET9 and RET51, which comprise 1,072 and 1,114 amino acids, respectively, are the main isoforms in vivo [17]. Animal studies have shown that RET9 is essential for kidney morphogenesis and enteric nervous development, while RET51 is essential for later kidney differentiation [19, 20].
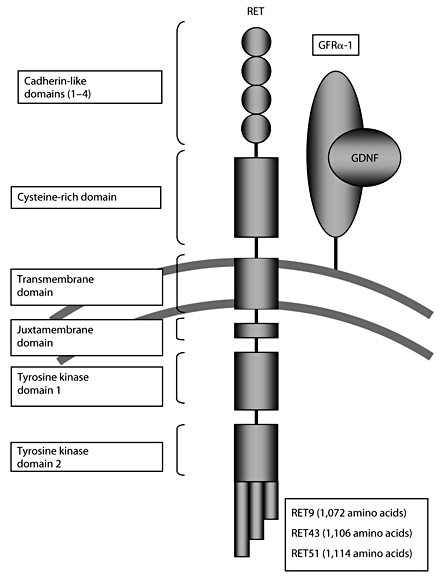
Schematic drawing of the RET protein with the 4 extracellular cadherin-like domains, the cysteine-rich domain, intracellular juxtamembrane domain and the intracellular tyrosine kinase domains. Three alternatively spliced forms, RET9, 43 and 51, are shown.
Schematic drawing of the RET protein with the 4 extracellular cadherin-like domains, the cysteine-rich domain, intracellular juxtamembrane domain and the intracellular tyrosine kinase domains. Three alternatively spliced forms, RET9, 43 and 51, are shown.
There are four ligands and coreceptors of the RET receptor. The ligands are the glial cell-line-derived neurotrophic factor (GDNF), neurturin, artemin and persephin [3]. RET is activated by each ligand through specific glycosyl-phosphatidylinositol-anchored coreceptors (GFRα1–4). The RET tyrosine kinase domain is a dimer under unstimulated conditions. It adopts a trans-inhibited head-to-tail inactive dimer conformation in which the substrate-binding site of each monomer is occluded by the contralateral one. Interaction of one of the 4 GDNFs with its specific GFRα coreceptor probably causes a conformational change that favours the formation of active dimers (RET dimerization, which leads to autophosphorylation at specific tyrosine residues in the intracellular domain), thereby relieving the trans-inhibition [21]. These phosphorylated tyrosines serve as docking sites for a number of transduction molecules which activate downstream signalling pathways. RET9 and RET43 contain 16 tyrosine residues, while RET51 has 2 additional tyrosines (Tyr1090 and Tyr1096) in the C-terminal end. Of these specific phosphorylation sites, Tyr1062 is crucially important in the activation of major intracellular signalling pathways [17]. Tyr1062 is a multidocking site for various adaptor proteins such as downstream of kinase (DOK) 1/4/5, Enigma, SH3 and multiple ankyrin repeat domains 3 (SHANK3), Src-homology collagen (Shc), ShcC (also named as Rai), fibroblast growth factor receptor substrate 2 (FRS2), insulin receptor substrate 1/2, ERK5, MAPK, phosphoinositide-dependent kinase 5 (CDK5), and PKC isoforms (fig. 2) [22]. The activation of Ras/ERK, phosphatidylinositol 3-kinase (PI3K)/Akt, p38 mitogen-activated protein kinase and c-Jun N-terminal kinase (JNK) pathways, which are predominantly responsible for cell proliferation and differentiation, occurs mainly through Tyr1062 [17]. The vital role of Tyr1062 phosphorylation in RET signalling and development was demonstrated by ‘knock-in’ transgenic mouse models in which the Tyr1062 residue of RET alleles was replaced by the ‘non-phosphorylatable’ amino acid phenylalanine [23]. These knock-in mice showed marked growth retardation and defects in enteric neurons and renal development, though this phenotype was less severe than complete RET knock-out mice.
![Schematic drawing of the RET signaling pathways. RET is activated by GDNF family ligands with glycosyl-phosphatidylinositol-anchored GDNF family receptor-α coreceptors. Major autophosphorylation sites are shown in circles with arrows to their direct targets. Tyrosine 752 (Y752) and Y928 binds STAT3, Y905 is a binding site for GRB7/10 adaptors, Y981 binds Src, Y1015 binds phospholipase Cγ, Y1096, unique to alternatively spliced form RET51, binds to GRB2 and Y1062 binds several proteins including DOK1, DOK4/5, Enigma, SHANK3, Shc, FRS2, SHC C, IRS 1/2. Potential therapeutic agents to block pathways are shown in boxes. RET = proto-oncogene RETprotein; RAC = Rho/RAC-family GTPase; SRC = proto-oncogene SRCprotein; FAK = focal adhesion kinase; PLCγ = phospholipase Cγ; IP3 = D-myo-inositol-1,4,5-trisphosphate; PKC = protein kinase C; DOK 1/4/5 = downstream of kinase; JNK = c-Jun N-terminal kinase; RAS = proto-oncogene RASprotein; RAF = proto-oncogene RAF protein; ERK = extracellular-signal-regulated kinase; MAPK = mitogen-activated protein kinase; MEK = MAPK or ERK kinase; SHANK3 = SH3 and multiple ankyrin repeat domains 3; Shc = Src-homology collagen; SOS = son-of-sevenless multi-protein scaffold complex; FRS2 = fibroblast growth factor receptor substrate 2; GAB1/2 = Grb2-associated binding protein; IRS1/2 = insulin receptor substrate; PI3K = phosphoinositide-3-kinase; AKT = protein kinase B; CREB = cAMP-responsive element-binding protein; p38 MAPK = p38 mitogen-activated protein kinase; IĸB = inhibitor of NF-ĸB; IKK = IĸB kinase; NF-ĸB = nuclear factor ĸB; PDK1 = phosphoinositide-dependent kinase 1; CDK5 = cyclin-dependent kinase 5; STAT = signal transducer and activator of transcription; JAK = Janus-activated kinase. Adapted from de Groot et al. [17].](https://karger.silverchair-cdn.com/karger/content_public/journal/nen/90/4/10.1159_000220827/3/m_000220827_f02.gif?Expires=1716908154&Signature=cNxg-ssKmAIeZsnK7DinW8psjFdfClN8ZMyznfUFlGHbazWcbavsHnhpRtvv-lUGypxK2kZ4UH8UVnoFQrm~zai9fvT1o2nkfL4WE4JIlrIN~bnIBxd539nFqGJ3R7zWGPz-aoJcSuAjY04RYgdNH5XHizeG8n~qIJm-EjKoTqItdF-AbHE41HyFGsqwNIuCbqm0k73Th~9-t4wVKMDm6EHTzcS64dC41TvXIhVWcbvjbN9R9qAYiOXlrbIkj3PF5QhfnW58QeKGYFA5nvEjkOJyiMZOTXlag8ZPeoRWFp5lDIL910qRtsAiAyGWeRCA~oAk6br8ff1TBa8jO16M0w__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Schematic drawing of the RET signaling pathways. RET is activated by GDNF family ligands with glycosyl-phosphatidylinositol-anchored GDNF family receptor-α coreceptors. Major autophosphorylation sites are shown in circles with arrows to their direct targets. Tyrosine 752 (Y752) and Y928 binds STAT3, Y905 is a binding site for GRB7/10 adaptors, Y981 binds Src, Y1015 binds phospholipase Cγ, Y1096, unique to alternatively spliced form RET51, binds to GRB2 and Y1062 binds several proteins including DOK1, DOK4/5, Enigma, SHANK3, Shc, FRS2, SHC C, IRS 1/2. Potential therapeutic agents to block pathways are shown in boxes. RET = proto-oncogene RETprotein; RAC = Rho/RAC-family GTPase; SRC = proto-oncogene SRCprotein; FAK = focal adhesion kinase; PLCγ = phospholipase Cγ; IP3 = D-myo-inositol-1,4,5-trisphosphate; PKC = protein kinase C; DOK 1/4/5 = downstream of kinase; JNK = c-Jun N-terminal kinase; RAS = proto-oncogene RASprotein; RAF = proto-oncogene RAF protein; ERK = extracellular-signal-regulated kinase; MAPK = mitogen-activated protein kinase; MEK = MAPK or ERK kinase; SHANK3 = SH3 and multiple ankyrin repeat domains 3; Shc = Src-homology collagen; SOS = son-of-sevenless multi-protein scaffold complex; FRS2 = fibroblast growth factor receptor substrate 2; GAB1/2 = Grb2-associated binding protein; IRS1/2 = insulin receptor substrate; PI3K = phosphoinositide-3-kinase; AKT = protein kinase B; CREB = cAMP-responsive element-binding protein; p38 MAPK = p38 mitogen-activated protein kinase; IĸB = inhibitor of NF-ĸB; IKK = IĸB kinase; NF-ĸB = nuclear factor ĸB; PDK1 = phosphoinositide-dependent kinase 1; CDK5 = cyclin-dependent kinase 5; STAT = signal transducer and activator of transcription; JAK = Janus-activated kinase. Adapted from de Groot et al. [17].
Schematic drawing of the RET signaling pathways. RET is activated by GDNF family ligands with glycosyl-phosphatidylinositol-anchored GDNF family receptor-α coreceptors. Major autophosphorylation sites are shown in circles with arrows to their direct targets. Tyrosine 752 (Y752) and Y928 binds STAT3, Y905 is a binding site for GRB7/10 adaptors, Y981 binds Src, Y1015 binds phospholipase Cγ, Y1096, unique to alternatively spliced form RET51, binds to GRB2 and Y1062 binds several proteins including DOK1, DOK4/5, Enigma, SHANK3, Shc, FRS2, SHC C, IRS 1/2. Potential therapeutic agents to block pathways are shown in boxes. RET = proto-oncogene RETprotein; RAC = Rho/RAC-family GTPase; SRC = proto-oncogene SRCprotein; FAK = focal adhesion kinase; PLCγ = phospholipase Cγ; IP3 = D-myo-inositol-1,4,5-trisphosphate; PKC = protein kinase C; DOK 1/4/5 = downstream of kinase; JNK = c-Jun N-terminal kinase; RAS = proto-oncogene RASprotein; RAF = proto-oncogene RAF protein; ERK = extracellular-signal-regulated kinase; MAPK = mitogen-activated protein kinase; MEK = MAPK or ERK kinase; SHANK3 = SH3 and multiple ankyrin repeat domains 3; Shc = Src-homology collagen; SOS = son-of-sevenless multi-protein scaffold complex; FRS2 = fibroblast growth factor receptor substrate 2; GAB1/2 = Grb2-associated binding protein; IRS1/2 = insulin receptor substrate; PI3K = phosphoinositide-3-kinase; AKT = protein kinase B; CREB = cAMP-responsive element-binding protein; p38 MAPK = p38 mitogen-activated protein kinase; IĸB = inhibitor of NF-ĸB; IKK = IĸB kinase; NF-ĸB = nuclear factor ĸB; PDK1 = phosphoinositide-dependent kinase 1; CDK5 = cyclin-dependent kinase 5; STAT = signal transducer and activator of transcription; JAK = Janus-activated kinase. Adapted from de Groot et al. [17].
It was also revealed that, upon ligand activation, RET was down-regulated by a pathway which was mediated by the Shc-Grb2 route which was activated through Tyr1062 and Tyr1096 [24].
RET Mutations and MTC
Activation of RET is both a growth and survival signal, and is a cause of MTC. There is experimental evidence demonstrating that RET mutations play a critical role in initiating MTC tumourigenesis. In a transgenic mouse model (CT-2A mice), RET51 cDNA was used to construct mice in which the most frequent MEN 2A mutation, Cys634Arg, was expressed under the control of the human calcitonin promoter [25]. These mice developed C cell tumours resembling human MTC and follicular tumours resembling human papillary thyroid carcinoma (PTC), depending on the founder line examined. In a model for MEN 2B, the human calcitonin gene (CALC-I) promoter was used to generate transgenic mice expressing the human RET oncogene with the MEN-2B-specific M918T mutation (CALC-MEN2B-RET) [26]. At 20–22 months of age, 3 out of 8 CALC-MEN2B-RET transgenic mice presented with macroscopic bilateral MTC. In 3 mice, nodular CCH was observed. In the control group, in which human non-mutated RETproto-oncogene was expressed in the C cells (CALC-WT-RET transgenic mice), thyroid abnormalities were never observed.
In human hereditary MTC, germline RET mutations are the genetic defect causing MEN 2A, MEN 2B and FMTC in around 98% of cases [21, 22, 23, 24, 25, 26, 27, 28, 29]. The mutated codons with their corresponding exons and the hereditary forms of diseases seen with them are shown in figure 3. In an analysis by the International RET Consortium in 1996, the most common (98%) mutations in MEN 2A were reported to be located in exons 10 and 11 of the RET proto-oncogene, encoding the highly conserved cysteine-rich domain [4, 21]. Codon 634 mutations are the most common among these, seen in 85% of MEN 2A patients. A single coding change (Cys634Arg) comprises 52% of all 634 mutations, and a second (Cys634Tyr) an additional 26% [9]. Less frequently, mutations involving tyrosine kinase domains have also been defined in MEN 2A. In FMTC, the most common (90%) mutations involve the extracellular, cysteine-rich domain but mutations involving the cadherin-like domain, and tyrosine kinase domains are also seen [4, 21]. Codon 634 mutations have been described in 30% of FMTC patients [4]. There are even mutations shown in only a single pedigree in FMTC [17]. In FMTC and MEN 2A double RET mutations, small insertions, deletions and small insertion-deletion mutations have been described as well [17]. MEN 2B is usually caused by a Met918Thr mutation in exon 16 in >95% of cases and an Ala883Phe mutation in exon 15 in 2–3% of cases, all in the tyrosine kinase 2 subdomain. Mutations can be de novo in some 50% of MEN 2B cases; thus, many patients with MEN 2B lack a family history [30].

Overview of the known germline mutations in the RET gene and their associated human diseases.
Overview of the known germline mutations in the RET gene and their associated human diseases.
Interestingly, some germline RET mutations (e.g., codon 804 mutations) may present with a variety of clinical courses, ranging between a late-onset and a relatively benign course to a more aggressive disease [30]. To present as MEN 2, these mutations either need to be in a homozygous state or, if they are heterozygous, they require a second germline or somatic mutation (or deletion) in RET, a downstream signalling gene or a tumour suppressor gene [31]. Thus, these mutations are believed to be of low transforming capacity and the impact of the second genetic alteration may explain the variability in clinical presentation [32, 33].
Although inherited mutations of RETlead to tumour formation in patients with MEN 2, it is not understood why only selected cells develop into tumours. In some previous studies performed in MTC tumour samples and TT cell lines (a human MTC cell line with a heterozygous codon Cys634Try RETmutation), allelic imbalance between mutated and wild-type RET as a possible mechanism for tumour formation was demonstrated [34, 35]. Activation of RET and subsequent tumour formation can occur by a ‘second hit’ causing a ‘dominant’ effect of the mutant allele: this may be due to loss of the normal wild-type allele, increased copy number of the mutant allele (e.g. duplication of the mutant allele as in trisomy 10), or tandem duplication of mutant RET[30].
In general, it can be stated that oncogenic tyrosine kinases such as mutant RET permit cells to function independent of their environment, to proliferate in the absence of growth factors, to protect against apoptosis, and to promote invasion and metastasis. Such a mutation also causes resistance to chemotherapy and a failure of cells to die, leading to the accumulation of genetic defects, which may explain their more aggressive behaviour; extracellular domain mutations of RET enhance growth. Normally, damaged DNA leads to cellular blockade at the G1/S checkpoint or, if damage is too great, to apoptosis. Activating mutations of RET promote allellic imbalance and prevent apoptosis, which cause abnormalities to accumulate.
Oncogenic RET activation and signalling is different from activation and signalling of non-mutated RET. In hereditary MTCs, the activation of oncogenic RET depends on the location of the amino acid change. In MEN 2A, where the mutations most commonly involve the extracellular cysteine-rich domain, a cysteine residue which is normally involved in an intramolecular disulphide bond is replaced by another amino acid causing disruption of the bond. This disruption causes the remaining cysteine partner to form an intermolecular disulphide bond causing RET dimerization and activation without ligand binding and leading to constitutive kinase activity [36]. However, close proximity to the RET transmembrane domain and the correct relative orientation of the cysteines are required for the disulphide bond to form. When the mutation occurs in the tyrosine kinase domain, as is generally seen in MEN 2B and FMTC, tyrosine residues in the kinase domain are activated; substrate binding specificity is changed secondary to structural changes of the binding pocket of the tyrosine kinase domain [17]. This leads to aberrant phosphorylation of substrates such as c-abl and c-Src, which does not occur with normal regulated ligand-binding. As a result, the mutated RETno longer requires dimerization to become active [37]. In a recent study, it was shown that codon 918 RET mutant (MEN 2B) is already active in the endoplasmic reticulum where it interacts with adaptor proteins [38]. Interestingly, several types of RET mutation can be further activated by GDNF as well [39].
One of the explanations for differences in transforming activity of RET receptors carrying different mutations is that these cause differences in cell surface expression of the mutated RET receptors. Codon 620 mutations are reported to cause enhanced constitutive homodimerization and consequent activation compared to codon 634 mutations [40]. Cell surface expression of RET receptors with codon 609, 611, 618 or 620 mutations has been shown to be very low compared with that of RET receptors with codon 634 mutation, suggesting that these former mutations impair transport of mutant RET receptors to the plasma membrane [41].
With regard to sporadic MTCs, somatic RET mutations in the tumour tissue of sporadic (non-familial) MTCs have been reported at different rates, in the range 40–50% [17, 42, 43]. Around 7–10% of apparently sporadic MTC are also found to have germline RETmutations; this is especially likely when MTC is multifocal or diagnosed at a younger age [44, 45]. The most common mutation is M918T in exon 16 followed by codon 804 and 768, but many other rare mutations have been described as well [17, 21]. However, RET mutations are not seen as a monoclonal change within individual MTC tissue samples, and they are not consistently distributed within primary tumours and metastases, suggesting that such mutations are not the sine qua non of carcinogenesis [46]. These mutations may occur during progression of the disease and probably contributeto disease phenotype instead of causing it. In a study by Elisei et al. [43], in which 100 sporadic-MTC patients were followed up for a mean of 10.2 years, it was revealed that only advanced stage at diagnosis and the presence of a RET mutation were independently correlated with a worse outcome. With regard to the effect of a somatic codon 918 RET mutation on survival in sporadic MTC, in 2 studies, M918T patients showed more aggressive development of distant metastases during follow-up with decreased metastasis-free survival, and they had a significantly lower survival rate than sporadic-MTC patients without the mutation [47, 48]. However, larger-scale studies are needed to validate the suggestion that somatic M918T mutations confer a high-risk status.
Although CCH precedes the development of inherited MTC, the role of CCH in sporadic MTC is less clear. It has been proposed by some authors that in sporadic MTC a single RET mutation is probably a sufficient event to cause CCH, which is the precursor lesion to MTC [42]. However, tumour progression after the mutation probably occurs as a result of clonal expansion caused by the accumulation of further somatic events. These may include chromosomal imbalance, which has been defined in more than half of the cases in some studies [49, 50], and mutations of other candidate genes.
RET Mutations and Downstream Signalling Pathways in MTC
There is little information available regarding the molecular mechanisms underlying the potent transforming and mitogenic potential of RET. Experimental models have been used to reveal the qualitative differences between signalling pathways when RET is activated by MEN 2A, FMTC or MEN 2B type mutations. In MEN-2-related RETmutations, a changing pattern and different levels of phosphorylation of docking sites (Y752, Y905, Y928 and Y1096) have been seen with different mutations [17]. Secondary to these differences in phosphorylation of docking sites, altered activation of downstream signalling is highly likely to occur; however, many of these pathways remain elusive. The main pathways involved, according to present evidence, are summarized below.
It was shown that, compared to RET activated by its natural ligand, the RET/MEN2A mutation impacts substantially on down-stream AKT signalling, while the RET/MEN 2B mutation more effectively activates both PI3K/AKT and JNK pathways which are responsible for survival signalling, enhanced cell-cycle progression and proliferation [51]. Some studies have shown possible changes in RET susbtrate specificity due to MEN 2B mutations, such as phosphorylation of paxillin and other proteins associated with the signalling proteins Crk and Nck compared to wild-type RET[52]. Adaptor proteins that preferentially bind to RET/MEN 2B compared to MEN 2A include Dok1, as well as JNK, which is strongly activated through its association with Dok1 [53]. In animal studies, it was demonstrated that the JNK pathway is involved in the ability of RET/MEN 2B cells to metastasize [54]. These findings may suggest that activation of the PI3K/AKT and JNK pathways might be responsible for the aggressive MTC phenotype in MEN 2B.
In a transgenic mice model with carcinomas carrying either RET/MEN 2A or RET/MEN 2B mutations, the expression of MAPK phosphatase-2 (MKP-2) was found to be induced [55]. MKP-2 is a dual-specificity phosphatase that selectively dephosphorylates p42/44 MAPK, p38 MAPK and JNK. Interestingly, despite MKP-2 inhibiting the activity of MAPKs, it was shown that depletion of MKP-2 results in deregulation of cell cycle and inhibition of proliferation in a RET/MEN 2A transgenic mouse carcinoma cell line [55]. In addition, the expression of MKP-2 was induced downstream of GDNF activation of normal RET, which may implicate its physiological role as a regulator of the duration and rate of MAPK activation. The authors of this study also reported increased expression of MKP-2 in MTCs of MEN 2A patients.
TT-raf cells are a human MTC cell line that contains an oestrogen-inducible raf-1 construct. It was previously shown that activation of raf-1 in TT-raf cells by oestradiol suppresses tumour cell growth and calcitonin secretion in vitro. In a further study, Vaccaro et al. [56]have utilized TT or TT-raf cells in a murine subcutaneous xenograft model to study tumour development and growth. Activation of raf-1 in mice with TT-raf tumours led to a significant decrease in MTC tumour formation. Control groups, however, had a high rate of MTC tumour development. These data indicate that raf-1 activation by estradiol treatment in this TT-raf xenograft model inhibited tumour development. Furthermore, to determine whether raf-1 activation could also inhibit the growth of established tumours, oestradiol and control pellets were implanted after tumour development. The TT-raf group that received oestradiol pellets showed an 8-fold decrease in tumour volume compared with the TT-raf control group. Taken together, these results suggest that in vivo activation of raf-1 in a murine model of MTC not only led to a reduction in tumour development, but also inhibited the growth of established tumours. These results suggest that strategies to activate the raf-1/MEK/ERK1/2 signalling pathway may be a viable approach to treat patients with metastatic MTC.
In a study performed in NIH-3T3 cells, FMTC mutations in RETcodons 791 and 891 induced constitutive STAT3 phosphorylation via a canonical Ras/ERK1/ERK2 pathway, and integration of Ras/ERK1/ERK2 and STAT3 pathways were required for up-regulation of the c-fos promoter by FMTC-RET [57]. In another cell line study, the same mutations were found to cause STAT3 activation through a pathway including Src, JAK1 and JAK2, independent of GDNF [58].
The cellular deposition of amyloid accompanying MTC is well known, and it has been shown that amyloid aggregation in neurodegeneration leads to hyperactivation of cyclin-dependent kinase 5 (Cdk5) and subsequent neuronal death. Based on the connection with amyloid, the role of Cdk5 in MTC was investigated [59]. Cdk5 inhibition by specific inhibitors or short interfering RNA decreased the proliferation of MTC cell lines, suggesting a novel and specific role for Cdk5 in supporting the proliferation of MTC cells.
In CALC-MEN2B-BRET transgenic mice which express a constitutively active oncogenic RET mutant, increased nuclear localization of β-catenin, which resulted in stimulation of β-catenin-specific transcriptional programmes in an RET-dependent fashion, was shown [60]. Also, in human MTC samples and metastases from MEN 2 patients with known oncogenic RET mutations, nuclear β-catenin expression was noted. However, this observation was made in a subset of cells and it was heterogeneously spread throughout the tumours. Nuclear localization of β-catenin was not detected in normal or hyperplastic C cells in mouse or human tissues. Together, these findings suggest that β-catenin signalling may play an important role in progression of RET-mediated tumours.
The nuclear factor-ĸB (NF-ĸB) family of transcription factors regulates a wide variety of cellular processes, including cell growth, differentiation and apoptosis. In a study by Gallel et al. [61], the immunohistochemical expression of members of the NF-ĸB family and the putative targets of NF-ĸB in a series of MTCs were assessed. Immunohistochemical evaluation included members of the NF-ĸB (p50, p65, p52, c-Rel, RelB) family, as well as putative targets of NF-ĸB such as Flip, Bcl-xL, and cyclin D1. Nuclear immunostaining for members of NF-ĸB was frequent in MTCs (p50, 19%; p65, 68%; p52, 86.6%; c-Rel, 75%; RelB, 36%). MTCs with germline or somatic RETmutations (29 cases) showed NF-ĸB nuclear translocation more frequently than in MTCs without RET mutations. Immunostaining for putative targets of NF-ĸB showed a significant statistical association between p65 and Bcl-xL. Bcl-xL expression was found to be statistically higher in the tumours with exon 16 RET mutation in comparison with those with exon 10 and 11 RET mutations or wild-type RET. To evaluate the significance of RET signalling in NF-ĸB activation, a RET-mutated TT cell line was infected with lentiviruses carrying short hairpin RNA to knockdown RETexpression, and NF-ĸB activity was assessed by luciferase reporter assays. Silencing of RET in the TT cell line produced a significant decrease in NF-ĸB activation and a reduction in ERK1/2. These results may suggest that the NF-ĸB is activated in MTCs, and supports the hypothesis that RET activation by somatic or germline mutations may be responsible for NF-ĸB activation in MTCs.
Mutations in SDH Genes in MTC
Genes encoding mitochondrial succinate dehydrogenase subunits B, C and D (SDHB, SDHCand SDHD genes) have also been investigated in sporadic and hereditary MTCs. In a study by Montani et al. [62], somatic mutations of the SDH subunits were absent in all 35 (22 sporadic, 13 MEN2-associated) MTCs investigated. An accumulation of amino-acid-coding polymorphisms was found among MTC patients, especially in patients with familial tumours, suggesting a possible role for SDH polymorphisms as disease-modifying factors in familial and sporadic MTC. In a study performed in 20 apparently sporadic MTCs without germline RET mutations, 30% of patients were found to have 5 different germline SDHB and SDHD mutations [63]. However, these apparent ‘mutations’ were also present in the normal, healthy population.In a study by Cascon et al. [64], in two families with hereditary non-RET CCH and in sporadic MTC cases, no association between SDHD mutations and hereditary CCH or sporadic MTC was found.
RET Polymorphisms and Haplotypes in MTC
A genetic locus is considered polymorphic if one or more of the rare alleles have a frequency of at least 0.01. Most polymorphisms do not alter the functional activity of the encoded protein, but not all polymorphisms are neutral. If the existence of a polymorphism or haplotype (a set of closely linked markers or polymorphisms inherited as a unit) associates with a certain phenotype, it might be that it acts as a genetic modifier and may be associated with a small increased relative risk of development of the disease. It is also possible that polymorphisms interact with other genetic variants and with germline MEN-2-associated mutations to affect development of features.
In MEN 2 and FMTC, several single-nucleotide polymorphisms (SNPs) and haplotypes of RET have been described in the general population [65] and and in association with MEN 2A [66]. In a study by Robledo et al. [66], the polymorphic G691S/S904S variant of RET was proposed to modify the age at which MEN 2A begins. However, these results were not replicated in a further study from the same center which included a larger patient group (n = 384) [67], or in another study from Spain [68]. In a further study, an association of the SNP L769L with the FMTC germline mutation F791Y was suggested [69].
RET polymorphisms and haplotypes have also been analysed in sporadic-MTC patients. However, the data regarding the differential distribution of RET germline SNPs among patients with sporadic MTC and controls are highly discrepant. The first major discordance among different studies was regarding the prevalence of each SNP: G691S ranged between 11.1 and 33%; L769L, 16 and 50%; S836S, 1 and 10% and S904S, 11.1 and 46% [69, 70, 71, 72, 73, 74, 75, 76]. Second, a polymorphism of exon 11 (G691S) has been reported to be more frequent in patients with sporadic MTC when compared with normal subjects [72, 75, 77], suggesting a possible role of this variant in RET activation.
In studies from the USA, Germany and Spain, SNP S836S was reported to be overrepresented and associated with the somatic mutation M918T in the tumoural DNA of sporadic-MTC patients [70, 78]. However, this polymorphism was not found to cause a predisposition to sporadic MTC in studies performed in French, Polish, British, Brazilian, Chilean, Portuguese or Austrian patients [69, 71, 73, 74, 75, 77].
In a study by Borrego et al. [79], the IVS1–126G→T polymorphism was shown to be significantly overrepresented in Spanish sporadic MTC patients, and the disease was associated with a specific haplotype within RETintron 1 that contains IVS1–126G→T and IVS1–1463T→C. In a further study, the same group studied 58 sporadic MTC patients compared with normal controls in order to identify a low-penetrance susceptibility locus for sporadic MTC in linkage disequilibrium with RET variants S836S/IVS1–126G→T [80]. IVS1–126G→T was again found to be overrepresented in sporadic MTC patients; however, no differences were obtained among cases and controls in the distribution of the variants tested upstream of this position. The frequency and distribution of G691S/S904S variants were also investigated in this study. Although G691S/S904S variants were previously found to be associated with sporadic MTC [32], and G691S was thought to be the functional polymorphism, G691S/S904S variants were similarly represented in both groups [80]. The association between this SNP (IVS-1–126G→T) and sporadic MTC was not demonstrated in a series of UK patients [77]. However, an association of a haplotype which includes the SNPs G691S, S904S and STOP + 388 bp on exon 19 and sporadic MTC was demonstrated [77]. With regard to germline sequence variant in intron 14 (IVS14–24G→A), it has been found to be both associated [69] and unassociated [81] with sporadic MTC. Although GFRα1–193, a polymorphism of the GFRα1 gene, was found to be associated with sporadic MTC in a small case-control study [82], this result was not confirmed in 2 larger studies [77, 83].
To identify low-penetrance genes related to the development of sporadic MTC, a two-stage case-control study in 2 European populations using high-throughput genotyping was performed in Spain [84]. In this study, 417 SNPs belonging to 69 genes either related to RET signalling pathway/functions or involved in key processes for cancer development were chosen. Tag SNPs and functional variants were included, where possible. These SNPs were initially studied in a large series of sporadic MTC cases (n = 266) and controls (n = 422), all of Spanish origin. In stage II, an independent British series of 155 sporadic-MTC patients and 531 controls was included to validate the previous results. Associations were assessed by an exhaustive analysis of individual SNPs but also considering gene- and linkage disequilibrium-based haplotypes. The authors identified 7 low-penetrance genes, 6 of which (STAT1, AURKA, BCL2, CDKN2B, CDK6, COMT) were consistently associated with sporadic-MTC risk in the 2 case-control series, and a 7th (HRAS) with individual SNPs and haplotypes associated with sporadic MTC in the Spanish data set. The potential role of CDKN2B was confirmed by a functional assay showing a role of a SNP (rs7044859) in the promoter region in altering the binding of the transcription factor HNF1.
In a very recent case-control study from Italy, a wide panel of 7 RET SNPs was tested in 140 sporadic MTC series and in a matched control group (n = 190) [85]. None of the investigated polymorphisms show a significantly different distribution in patients with sporadic MTC when compared to controls. Twenty haplotypes and 57 genotypes were generated, and their association with the disease and the clinical features was statistically evaluated. Interestingly, 14 genotypes were found to be unique to sporadic-MTC patients and 25 to controls. Two haplotypes and 3 genotypes, all including the intronic variants IVS1–126 and IVS14–24, were significantly associated with sporadic-MTC patients and with a higher tumour aggression. The functional activity of the only non-synonymous RET variant (c.2071C→A, G691S) was tested for the first time. Interestingly, Western blot analyses showed that the fraction of Ret9-G691S protein located at the plasma membrane level was overrepresented when compared to Ret9-WT, suggesting facilitated targeting at the cell membrane for this variant. However, no transforming activity was shown in a focus-formation assay on cells carrying the Ret9-G691S, against a possible oncogenic role of G691S variant.
In summary, while many interesting associations of SNPs have been related to MTC, in most cases these were not confirmed in other or larger populations. The utility of this approach remains unclear at present.
Gain-of-Function Mutations of Proto-Oncogenes and Loss-of-Function Mutations of Tumour Suppressors in MTC
To date, gain-of-function mutations in known proto-oncogenes such as RAS and BRAF in MTC have been reported in only one study, from Greece. A K-RAS codon 12 mutation was found in 41% and the BRAF V600E mutation in 68% of the MTC samples [86].
There are 3 families of tumour suppressor genes: pRb (retinoblastoma protein), tumour protein p53, and phosphatidylinositol-3,4,5-triphosphate 3-phosphatase and dual-specificity protein phosphatase (PTEN). No loss-of-function mutations in any of these genes have been identified in MTC.
During cell cycle progression, the critical transition from the G1 to S phase is controlled by a family of serine-threonine protein kinases, collectively referred to as cyclin-dependent kinases (CDKs). CDKs are negatively regulated by CDK inhibitors (CDKIs). CDKIs are grouped into 2 families – INK4 and CIP/KIP. Several reports have shown that p18 from the INK4 family and p27 from the CIP/KIP family cooperate in cell cycle arrest of different cell types [87]: p18 –/– mice develop MTC, but the incidence is enhanced by the additional loss of p27 [88], while p27 –/– mice develop MEN-like tumours only in combination with loss of p18 [89]. This suggests that p18 and p27 collaborate in suppression of tumourigenesis, and both are regulated by RET. In a cell line expressing a MEN-2A-specific codon 634 RET mutation, it was demonstrated that p18/p27 activity was reduced and cyclin D protein levels were elevated leading to increased CDK activity and increased phosphorylation of pRb. Proliferation was seen in these cells under growth arrest conditions [89]. Blocking RET 2A-MAPK signals with MAPK kinase (MEK) inhibitors re-established normal elevated p18/p27 expression and growth arrest, but did not affect the inappropriately elevated levels of D-type cyclins which implicates the MAPK pathway in RET 2A-mediated p18/p27 changes but not in cyclin D regulation. It was previously shown that 13% of transgenic mice expressing the human RET MEN 2B mutation develop MTC at 11–24 months of age whereas CCH was seen in 77% of these mice from 8 months onward [26]. The incomplete penetrance and variable latency period for MTC development in these mice have led to the idea that other tumourigenic events are required for the development of MTC. Van Veelen et al. [87]have reported several somatic inactivating mutations in p18 in a subset of human MTCs, indicating that p18 is a tumour suppressor gene which may be involved in human MTC development. Based on this finding and previous studies, a transgenic mouse model was developed in which mice expressing oncogenic RET2B were crossed with mice lacking p18 and p27 and monitored for MTC development [87]. RET2B;p18+/– mice and RET2B;p18–/– mice developed MTC with a highly increased incidence compared with their corresponding single-mutant littermates. Heterozygous loss of p27 did not result in increased MTC incidence in RET2B mice up to 12 months of age. This indicates that loss of p18, but not loss of p27, cooperates with oncogenic RET in MTC development.
Human FHIT (fragile histidine triad) gene is a highly conserved gene whose loss of function may be important in the development and/or progression of various types of cancer. In a study by Pavelic et al. [90], FHIT and p53 gene status in different benign and malignant thyroid tumours including MTC was investigated. Eight of 15 MTCs harboured single point mutations or loss of heterozygosity: 68% of MTC samples showed negative FHIT protein expression, while 7 MTC samples displayed aberrant expression of the FHIT gene. The results showed a correlation between aberrant FHIT and p53 expression, a low rate of apoptosis, and malignancy. Concomitant aberration of FHIT gene and p53 might be responsible for development of highly malignant types of thyroid cancer including MTC.
Other Possible Mechanisms
In some cancers such as prostate, ovarian, breast and colon, loss of oestrogen receptor-β (ERβ) expression was reported to play a role in progression of the cancer. In tumour versus normal tissue studies, expression of ERβ mRNA and protein decreased or the ERα/ERβ mRNA ratio increased in these cancer tissues [91]. This issue has also been studied in a few studies in the literature on MTC. In a limited study performed in 11 human MTC tissue samples, ERα was detected in 10/11 samples and ERβ was expressed in 3/11 [91]. The presence of mRNA for ERα and ERβ has also been demonstrated in human MTC tumour tissues [92]. In a study by Blechet et al. [93], ERβ was expressed in CCH in 100% and in MTC 96.5% of cases whereas ERα was never expressed. Thus, the hypothetical role of ER subtypes in human MTC remains to be established.
Maintenance of telomere length has been reported to be an absolute requirement for unlimited growth of human tumour cells: in 85% of cases this is achieved by reactivation of telomerase, the enzyme that elongates telomeres [94]. MTC is a rare example of human tumours in which telomerase activity is low or undetectable. There is only a single study in the English literature presenting evidence that even this low telomerase activity is sufficient to increase the replicative life span of human MTC cells [94].
Previous studies have demonstrated that cyclooxygenase-2 (COX-2) expression is associated with the carcinogenesis of numerous neoplasms. The expression of COX-2 in MTC was evaluated in an immunohistochemical study by Popovtzer et al. [95]. They evaluated tissue specimens of 22 patients with MTC and 15 control subjects with non-malignant thyroid tissue, concluding that COX-2 is overexpressed in the MTC tissue. These findings may have important treatment implications for the use of COX-2 inhibitors in patients with MTC.
In a study by Santarpia et al. [96], which investigated the mechanisms of sporadic MTC other than RET mutations using high-resolution array-based comparative genomic hybridization, allelic loss was defined as the predominant genomic abnormality found in MTC. The same group evaluated the allelic loss of 152 genes involved in the major pathways for DNA repair in 20 sporadic MTC tumour samples, 10 with activating somatic RET mutations [97]. A core set of 10 genes showed consistent allelic loss in greater than 25% of the tumours. The most frequent losses were observed for XAB2 (nucleotide excision repair) and CHAF1A (maintaining of chromatin structure), both occurring in 40% of the tumours. For tumours with somatic RETmutations, this frequency was 100%. Of particular interest was the observation that allelic loss of DNA repair genes was associated with metastasis.
In one study, the frequency of mitochondrial DNA mutations in MTC was investigated [98]. In this study, in 26 MTC tumour specimens (13 sporadic and 13 familial MTC) and their matched normal tissues, mitochondrial DNA mutations were analysed by sequencing the entire coding regions of the mitochondrial genome. Non-synonymous mutations were detected in 20 MTC samples (76.9%): 9 out of 13 sporadic MTC (69.2%) and 11 out of 13 (84.6%) familial MTC/MEN 2. Both transition and transversion types of mutations were found in the samples. Interestingly, 76.2% of transversion mutations were found in FMTC/MEN 2 patients whereas 66.7% of transition mutations were in sporadic MTC. Synonymous mutations were found in 12 MTC samples. In total, 27 transversion mutations (21 non-synonymous and 6 synonymous) were found. Of them, 81.5% were from FMTC/MEN 2, and 18.5% were from sporadic MTC. The association of transversion mutation with familial MTC/MEN 2 was statistically significant. The majority of the mutations were involved in the genes located in complex I of the mitochondrial genome, and often resulted in the change of a moderately or highly conserved amino acid in their corresponding protein. Mitochondrial respiratory function was also compromised in a TT cell line, which carries mtDNA mutations at nt 4917 and 11,720, and in peripheral lymphocytes of MTC patients with mtDNA mutations. These data may suggest that mitochondrial DNA mutations may be involved in MTC tumourigenesis and progression.
Pituitary tumour transforming gene-1 (PTTG-1), a mammalian securin also exhibiting oncogenic properties, is overexpressed in a variety of endocrine-related tumours. In a very recent study by Zatelli et al. [99], PTTG-1 expression in human CCH, human MTC and the human MTC TT cell line was investigated. Moreover, the role of PTTG-1 in neoplastic C-cell proliferation was studied by investigating effects of specific PTTG-1 gene silencing. PTTG-1 mRNA expression was found to be significantly higher (p < 0.01) in CCH (threefold) and in PTC and MTC samples (fivefold) than in normal thyroid specimens and in MTC lymph node metastases as compared to primary thyroid lesions. Moreover, PTTG-1 mRNA expression correlated with tumour diameter and TNM status. Similar results have been demonstrated for PTTG-1 protein. In TT cells, PTTG-1 silencing by siRNA nucleofection did not completely block DNA synthesis, but caused a significant reduction in [3H]thymidine incorporation, as compared to TT cells transfected with GAPDH siRN. The reduced cell proliferation rate persisted up to 6 days, supporting the hypothesis that PTTG-1 may play an important role in C cell neoplastic proliferation.
Novel Treatment Modalities: Preclinical Studies
Drugs under Investigation
As currently the only effective treatment modality for MTC is surgery, drug therapy for MTC has been an area of intensive research. There are a number of different drugs and treatment approaches which act on different pathways and which have been or are still being investigated in cell lines and animal models.
Bortezomib is a proteasome inhibitor and its mechanism of action is complex, but appears to include the inhibition of inhibitory-ĸB (IĸB) degradation, leading to the accumulation of IĸB and hence inactivation of NF-ĸB. MTC show high levels of NF-ĸB (see above), and in a study in human MTC cell lines, bortezomib induced apoptotic cell death with IC50 values well within the range of clinically achievable concentrations [100]. Bortezomib also induced increased sensitivity of MTC cells to low-dose doxorubicin. These findings may be promising for future clinical trials.
Lithium chloride is a potential agent which was shown to inhibit glycogen kinase synthase-3-β (GSK-β), a downstream target of the raf-1 pathway. In a study in a TT cell line and in TT xenograft mice, lithium chloride was found to be effective in inhibiting growth in the cells while LiCl-treated TT xenograft mice showed a significant reduction in tumour volume compared with controls [101].
Histone deacetylases (HDACs) and histone acetyltransferases exert opposing enzymatic activities that modulate the degree of acetylation of histones and other intracellular molecular targets, thereby regulating gene expression, cellular differentiation, and survival. Two HDAC inhibitors, suberoylanilide hydroxamic acid (SAHA) and m-carboxycinnamic acid bis-hydroxamide, have been tested in MTC cell lines [102]. Both drugs induced growth arrest and caspase-mediated apoptosis; SAHA down-regulated the expression of the apoptosis inhibitors FLICE-like inhibitory protein (FLIP) and cellular inhibitor of apoptosis-2 (cIAP-2), and sensitized tumour cells to cytotoxic chemotherapy. Recent research has also shown that the Notch signalling pathway is regulated by an HDAC corepressor complex that is sensitive to HDAC inhibitors [103]. Thus, in another study suberoyl bis-hydroxamic acid (SBHA) was shown to activate Notch-1 signalling, which was associated with antiproliferative and apoptotic effects on MTC cells [103]. Valproic acid is an anti-epileptic agent which has recently been shown to be a potential Notch 1 activator: as Notch 1 overexpression inhibits MTC cell growth and hormone production, Greenblatt et al. [104]went on to show that valproic acid activates Notch1 signalling in MTC cells and inhibits their growth by inducing apoptosis, which may thus make it a readily available potential treatment agent for MTC.
Preclinical studies have also examined the effect of RET inhibition in combination with cytotoxic drugs. In a preclinical model, the cytotoxic agent and topoisomerase I inhibitor irinotecan was tested alone or in combination with an indolocarbazole derivative and tyrosine kinase inhibitor CEP-751 in TT cell lines or TT cell xenografts in nude mice [105]. In TT cell culture and xenografts, irinotecan treatment was highly effective and this effect was augmented by treatment with CEP-751. Treatment of TT cell xenografts resulted in durable complete remission in 100% of the mice, with a median time to recurrence of 70 days for irinotecan alone and more than 130 days for irinotecan plus CEP-751 for 5 of 9 treated mice. Currently, a phase II trial is recruiting MTC subjects for testing irinotecan (table 1). The indolocarbazole derivatives CEP-701 and CEP-751 inhibited RET autophosphorylation and proliferation of TT cells, while CEP-751 inhibited tumour growth in nude mice that have been injected with TT cells [106].
Current open clinical trials for MTC

Tagliati et al. [107] and Zatelli et al. [108, 109] have studied somatostatin analogues in MTC cell lines in vitro in several studies. Somatostatin was shown to reduce cell growth in the human MTC cell line TT, which expresses all somatostatin receptors. The inhibitory effect of somatostatin on cell proliferation was at least in part mediated by a cytoplasmic tyrosine phosphatase, Src homology-2-containing protein (SHP)-1, which in turn is activated upon somatostatin binding to SSTR2. They also showed that SHP-1 activation downregulates MAPK signalling in TT cells, resulting in decreased cell proliferation. In one of their studies they investigated 18 human MTC tumour samples expressing SSTR1, SSTR2 and SSTR5 [108]. Cell viability and calcitonin secretion were investigated in primary cultures of these MTC samples. The cultures were divided into two groups according to the extent of calcitonin secretion inhibition after treatment with BIM-23014 (lanreotide). Cultures responding to lanreotide with a calcitonin reduction of 15% or greater versus untreated cells were considered as responders. According to this, 9 tumours were responders and 9 were non-responders. In the ‘responder’ group, calcitonin secretion was reduced by compounds interacting with SSTR1, SSTR2 and SSTR5 whereas cell viability was not affected. On the other hand, in the ‘non-responder’ group, calcitonin secretion was reduced by an SSTR1 selective agonist whereas cell viability was inhibited by SSTR2-selective agonists [108].
Chemotherapy failure has been ascribed, at least in part, to the overexpression by MTC of the multidrug resistance 1 (MDR1) gene, encoding a transmembrane glycoprotein [permeability glycoprotein (P[-]gp)] that antagonizes intracellular accumulation of cytotoxic agents. P-gp expression and function in a rat model have been demonstrated to depend on cyclooxygenase (COX)-2 isoform levels, which as noted above, may be overexpressed in MTC (as in some other cancers). In another study, Zatelli et al. [110] investigated the role of the COX-2 pathway in modulating chemoresistance, and then evaluated the sensitizing effects of COX-2 inhibitors on the cytotoxic effects of doxorubicin in the presence or in the absence of prostaglandin E2 in primary cultures and in human MTC cell line, TT. It was shown that TT cells express both MDR1 and COX-2 and that rofecoxib, a selective COX-2 inhibitor, sensitizes TT cells to the cytotoxic effects of doxorubicin, reducing P-gp expression and function. In 3 other studies investigating individual SST receptor subtype effects on the TT cell line, they showed that SSTR2 activation inhibits DNA synthesis and cell proliferation whereas SSTR5 activation increases DNA synthesis: somatostatin reduces cell proliferation, calcitonin secretion and calcitonin gene expression [111], selective activation of SST1 with agonist drugs BIM-23926 and BIM-23745 significantly reduces calcitonin secretion and gene expression with a reduction in CREB phosphorylation [112]. In another study, it was reported that in a tumoural TT cell line, indomethacin in vivo and in vitro decreases proliferation via 15-PGDH, the PG catabolism key enzyme [113].
NVP-AST487, a RET kinase inhibitor, was investigated in NIH3T3 and TT cell lines by Akeno-Stuart et al. [114]. NVP-AST487 induced a dose-dependent growth inhibition of xenografts of NIH3T3 cells expressing oncogenic RET, and of the MTC cell line TT in nude mice. Furthermore, NVP-AST487 was found to inhibit calcitonin gene expression in vitro in TT cells, in part through decreased gene transcription. RPI-1, a 2-indolinone RET tyrosine kinase inhibitor, was also tested on cells that express RET C634 oncogenic mutants common in the MEN 2A syndrome. NIH3T3 fibroblasts were transfected with RET(C634R) while TT cells express endogenous RET(C634W) [115]. In NIH3T3 cells expressing the RET mutant, RET protein and tyrosine phosphorylation were undetectable after 24 h of RPI-1 treatment. In TT cells, RPI-1 inhibited proliferation, RET tyrosine phosphorylation, RET protein expression, and the activation of PLCγ, ERKs and AKT. In mice, oral daily RPI-1 treatment inhibited the tumour growth of TT xenografts by 81% and reduced the plasma levels of the specific biomarker calcitonin: 25% of RPI-1-treated mice were tumour free. In another study, the efficacy and the cellular basis of antitumour activity of the RET tyrosine kinase inhibitor RPI-1 was investigated against large established subcutaneous TT tumour xenografts in mice [116]. Oral treatment with RPI-1 caused growth arrest or regression in 81% of treated tumours. Following treatment suspension, tumour inhibition was maintained and ‘cures’ were achieved in 2/11 mice. These findings indicate that the antitumour effect of RPI-1 on the MTC is characterized by apoptosis induction and angiogenesis inhibition.
As RET stimulates several intracellular signalling cascades after activation, inhibition of these pathways has also been studied as potential targets. PI3K inhibition by LY294002 was shown to suppress growth in MTC by increasing apoptosis [117]. In a study by Ludwig et al. [118], inhibition of constitutive NF-ĸB activity was shown to result in cell death of TT cells and block focus formation induced by oncogenic forms of RET in NIH-3T3 cells. These results suggested that RET-mediated carcinogenesis may depend on IĸB activity and subsequent NF-ĸB activation.
Other growth factor receptor inhibitors, apart from those with RET kinase blocking activity, have been evaluated as a treatment option in the inhibition of different cell lines [119, 120, 121]. PD173074, a fibroblast growth factor receptor (FGF-R) inhibitor, resulted in abrogation of fibroblast growth factor-1-mediated FGF-R4 phosphorylation in TT cells, an effect that was accompanied by significant arrest of cell proliferation and tumour growth in vivo [119]. PP1, a pyrazolopyrimidine, was reported to be an efficient c-Src-related kinase and PDGF-R inhibitor: it inhibited RET/PTC3 autophosphorylation in a dose-dependent manner in NIH3T3 cells expressing the RET/PTC3 oncogene [121]. PP1 also blocked anchorage-independent growth and tumourigenicity in nude mice of NIH3T3 fibroblasts expressing the RET/PTC3 oncogene. Another pyrazolopyrimidine, PP2, prevented serum-independent growth of RET/PTC1-transformed NIH3T3 fibroblasts in a study by the same group [122]. In a further study, the sensitivity of two human MTC cell lines to novel pyrazolopyrimidine derivates, which are able to inhibit src family tyrosine kinase activity, was assessed [123]. In TT cells carrying the multiple endocrine neoplasia MEN 2A RET mutation C634T, and in MZ-CRC-1 cells carrying the MEN 2B RET mutation M891T, the agent Si34 significantly inhibited growth. In a more recent study by Croyle et al. [120], EGF-R kinase inhibitors PKI166, gefitinib and AEE788 inhibited cell growth induced by various constitutively active mutants of RETin NIH3T3 cells and in the human PTC cell line TPC1, which harboursa naturally occurring RET/PTC1 rearrangement. Additionally, PKI166 inhibited cell growth of medullary carcinoma cell line TT cells. These data may indicate that EGF-R contributes to RET kinase activation, signalling and growth stimulation, and may therefore be an attractive therapeutic target in RET-induced neoplasms. SU5416 (semaxanib) is a potent inhibitor of vascular endothelial cell growth factor receptor (VEGFR), stem cell factor receptor (c-Kit) and Fms-like tyrosine kinase-3 receptor (FLT-3) receptor tyrosine kinases. In a study by Mologni et al. [124], SU5416 was shown to inhibit RET with similar potency, both in cell-free assays and in cells, and caused proliferation arrest in oncogenic RET-transfected cells and in papillary thyroid carcinoma (PTC) cells expressing the RET/PTC1 oncogene, but not in RET-negative control cells. SU5416 inhibited RET-mediated signalling through the extracellular signal regulated kinase (ERK) and JNK pathways. In addition, they have shown that although a naturally occurring MEN2 mutation at codon 804 confers resistance to SU5416, there was no resistance to the related compound SU4984. Substituted 4-(3-hydroxyanilino)-quinoline compounds, initially identified as small-molecule inhibitors of src family kinases, have been reported as potential inhibitors of RET kinase in one study [125]. Three compounds, 38, 31, and 40, were proposed as potential treatment agents in MTC, but at present they require further study.
The effect of gemcitabine, an agent which inhibits DNA synthesis and repair, on proliferation and the neuroendocrine activity of human TT cell line was investigated [126]. A concentration-dependent inhibitory influence of gemcitabine on the proliferation of TT cells was observed.
Members of the heat shock protein family function as molecular chaperones, ensuring the proper folding of newly translated proteins. Heat shock protein 90 (hsp90) mediates the folding of a limited number of client proteins, including steroid hormone receptors and signalling kinases. In a study by Marsee et al. [127], hsp90 and its co-chaperone p50cdc37were demonstrated to be novel proteins associated with RET/PTC1. RET/PTC1 decreases expression of the sodium/iodide symporter (NIS), the molecule that mediates radioiodide therapy for thyroid cancer. Inhibition of hsp90 function with 17-allylamino-17-demethoxygeldanamycin (17-AAG) was shown to reduce RET/PTC1 protein levels. Furthermore, 17-AAG increased radioiodide accumulation in thyroid cells by decreasing iodide efflux. Finally, the ability of 17-AAG to increase radioiodide accumulation was not restricted to thyroid cells expressing RET/PTC1. While most relevant to non-MTC thyroid neoplasia, these findings suggest that 17-AAG may be useful as a chemotherapeutic agent for MTC in the future.
Activation of phosphatases leads to the accelerated removal of substrate phosphates which could counter kinase signalling [17]. Several protein phosphatases have been shown to dephosphorylate RET and interrupt RET signalling, even in some activated RET mutants. Ptprj is a transmembrane tyrosine phosphatase that can dephosphorylate VEGF-Rs, PDGF-Rs, Met and oncogenic and wild-type RET [128]. However, although this phosphatase was found to be effective against the MEN 2A mutant form C634R, MEN 2B RET M918T was not affected by it. Another transmembrane tyrosine phosphatase, LAR/PTPRF, was able to reduce the oncogenic activity of MEN 2A RET but not MEN 2B RET [129]. The overexpression of a cytoplasmic tyrosine phosphatase, SHP1, which dephosphorylates RET at Y1062, was associated with growth inhibition in MTC cells [130]. As mentioned previously, SHP1 is activated by somatostatin receptor SSTR2 [109].Thus, activation of these phosphatases may be useful molecular targets for future treatment.
In a cell line study by Ludwig et al. [118], it was shown that inhibition of NF-ĸB activity could be a useful tool in the future for treatment of tumours with certain mutation types of RET. TT cells were shown to display transcriptionally active RelA(p65) in the nucleus; NF-ĸB activity in these cells was attributable to constitutive IĸB kinase (IKK) activity and high turnover of IĸB-α. RET harbouring the mutations C634R (MEN 2A) or M918T (MEN 2B), in contrast to wild-type RET, activates a NF-ĸB-dependent reporter construct upon transient transfection in HeLa cells. It was shown that the prototype RET mutation C634R enhances phosphorylation of IĸB-α by IKK-β but not by IKK-α. RET-induced NF-ĸB and IKK-β activity required Ras function and was dependent on Raf and MEKK1. Inhibition of constitutive NF-ĸB activity resulted in cell death in TT cells and blocked focus formation induced by oncogenic forms of RET in NIH 3T3 cells. These results may suggest that RET-mediated carcinogenesis critically depends on IKK activity and subsequent NF-ĸB activation.
A novel approach has been to block RET with specific oligonucleotide ligands called aptamers. These aptamers can be used to identify markers on the surface of a cell type and allow in vivo targeting for diagnostic and therapeutic applications. Aptamers are specific, they have a high affinity for their target, and they are poorly immunogenic. Aptamers can be generated for a variety of targets through evolution of a random pool of sequences (SELEX). In a study by Cerchia et al. [131], nuclease-resistant aptamers that recognize the human receptor tyrosine kinase RET were obtained using RET-expressing cells as targets in a modified SELEX procedure. One of these aptamers blocked RET-dependent intracellular signalling pathways by interfering with receptor dimerization when the latter was induced by the physiological ligand or by an activating mutation. It was speculated that this strategy could open the way to a new type of treatment, but currently its efficacy needs to be established.
Survivin is a novel member of the inhibitor of apoptosis protein (IAP) family which is known to be overexpressed in various carcinomas and associated with their biologically aggressive characteristics. In a study by Du et al. [132], immunohistochemical analysis showed high survivin expression in MTC and TT cells whereas no immunoreactivity was detectable in normal thyroid tissue. Statistical analyses revealed no significant correlation of survivin expression with the clinicopathologic features of MTC. In TT cells, survivin expression at both mRNA and protein levels was confirmed and could be downregulated by antisense oligodeoxynucleotides (ASODNs) concomitant with a decrease in viability and growth, and increase in apoptosis. These results may suggest that survivin plays an important role in MTC independent of the conventional clinicopathologic factors, and ASODNs are a promising survivin-targeted gene therapy for MTC.
The EGF-7TM membrane protein CD97 is a dedifferentiation marker in undifferentiated thyroid carcinomas and MTC. Betulinic acid (triterpenic acid) induces apoptosis in various epithelial and mesenchymal malignoma cell lines. The effects of betulinic acid (BA) and the betulinic acid butyrate ester (BABE) in two human MTC cell lines, MTC-SK and MTC-TT, were evaluated: both showed inhibition of proliferation [133]. Further studies need to be performed on protein and RNA-chip level to clear the question whether both substances are potential antitumour agents in MTC.
Another approach has been to generate molecular mimetics directed towards specific mutations of the REToncogene [134]. Substitution of Cys-634 in the extracellular domain of the RET tyrosine kinase receptor causes dimerization and activation of its transforming potential. In this study, a mutant protein consisting of the entire ectodomain of the RET carrying a Cys634Tyr substitution (EC-RetC634Y) was purified. The protein was glycosylated, as the native one, and was biologically active. By using an in vitrocell system, it was shown that EC-RetC634Y inhibits membrane-bound receptor RetC634Y, interfering with its dimerization. Furthermore, it was demonstrated that EC-RetC634Y competes with the wild-type RET receptor for ligand binding.
Targeting RET with a monoclonal antibody might also be therapeutically useful [135, 136, 137]. In a study by Stein et al. [137], the effect of labetuzumab, a humanized anti-CEA monoclonal antibody, on the growth of MTC, alone and in combination with chemotherapy, was investigated. Antitumour effects were evaluated in a nude mouse-human MTC xenograft model. Labetuzumab was found to have direct, specific, antitumour effects in that model, without conjugation to a cytotoxic agent. In addition, labetuzumab sensitized these tumour cells to chemotherapy (DTIC in this model) without increased toxicity. Significant delays in tumour growth were caused by the monoclonal antibody therapy or chemotherapy alone; however, the combination of these agents was significantly more effective than either agent alone.
Immunotherapy using autologous dendritic cells to induce an antigen-specific Th1-driven immunity has been attempted in MTC patients. While the results of these few studies have been encouraging, it is not possible as yet to draw any long-term conclusions regarding this approach [138].
Finally, gene therapy has been investigated in MTC cells. These can be categorized under 4 groups: (1) corrective gene therapy using dominant-negative RETmutants or ribozymes to inhibit REToncogene activity; (2) cytoreductive gene therapy using prodrug-activating enzymes, toxin genes or the sodium iodide symporter (SLC5A5) gene to allow 131I uptake; (3) immunomodulatory gene therapy, for example using cytokines; and (4) combined approaches that use cytoreductive plus immunomodulatory gene therapy [139]. The delivery of these gene therapies has primarily used viral vectors derived from human adenovirus serotype 5. Results from preclinical in vitroand in vivo studies are encouraging, but no gene therapy has yet been carried through to phase I clinical studies in patients with MTC.
Chemotherapy
While in general conventional chemotherapy has not been shown to be particularly effective in MTC, some preclinical and small clinical studies have shown promising results. In a study by Matuszczyk et al. [140], 9 patients (mean age 51 years) with MTC were treated with doxorubicin with 2 regimens: either 8 cycles of 15 mg/m2 weekly or 3 cycles of 60 mg/m2 every 3 weeks, repeated once, depending on response and side effects. The effect of therapy was evaluated by radiographic imaging, [18F]-FDG-PET, and bone scans: 11% showed partial regression over 6 months followed by stable disease for 3 months, 11% had stable disease over 7 months, while 79% demonstrated progressive disease established over 5 months (range: 2–12). In another study, combinations of doxorubicin and streptozocin and 5-FU and dacarbazine were given alternately to 20 patients with metastatic MTC [141]: 3 partial responses and 10 with long-term stabilization were observed. In a study by Wu et al. [142], 7 patients with advanced MTC were treated with cyclophosphamide (750 mg/m2), vincristine (1.4 mg/m2), and dacarbazine (600 mg/m2 daily for 2 days in each cycle) every 3 weeks. Two patients had partial tumour and biochemical responses for durations of 14 and 29 months, respectively. One patient had a partial biochemical response and stable tumour measurements for 9 months, and another patient had stable tumour size and markers for 14 months; 3 patients had progressive disease. Diarrhoea and flushing improved in 2 patients who had partial biochemical responses. One problem with these trials is that none of them used the Response Evaluation Criteria in Solid Tumour (RECIST) guidelines, which makes the interpretation of results difficult. Combretastatin is a tubulin inhibitor whose metabolite selectively inhibits proliferating cells in tumours [143]. In a preclinical model, it was shown that the anti-neovascular agent combretastatin A-4 phosphate prodrug (CA4P) in combination with doxorubicin was effective in curtailing tumour growth [144]. This combination of combretastatin and doxorubicin extended the doubling time of established MTC tumours in nude mice to 29 days, compared to 12 days in untreated controls. However, there are no further studies available regarding this treatment regimen. In a human MTC cell line study, camptothecin and paclitaxel were evaluated [145]. Although promising results were provided, these drugs have not so far been used in patients with MTC. There are also a few reports regarding chemotherapy treatment in individual patients with MTC. Plitidepsin is a new agent whose primary mechanism of action in tumour cells is the subject of ongoing investigation. Plitidepsin induces an early oxidative stress response, which results in a rapid and sustained activation of the EGF-R, the non-receptor protein tyrosine kinase Src, the serine threonine kinases c-Jun NH2-terminal kinase, and p38 mitogen-activated protein kinase. These early events rapidly trigger the induction of the mitochondrial apoptotic pathway via cytochrome c release, activation of the caspase cascade, and activation of protein kinase C, which seems to exert an important effector role in mediating cellular death induced by the drug [146]. In a phase I clinical trial, the single MTC patient showed disease stabilization [146]. Further studies with large patient groups are needed to evaluate the drug’s effect on MTC. Capecitabine, a thymidylate synthase (TS) inhibitor, has been used in MTC: Labidi et al. [147] reported a single case with metastatic MTC in progression after primary treatment with cisplatin-doxorubicin who showed prolonged remission with capecitabine, as did Paiva et al. [148].
Aplidine is a marine cyclodepsipeptide and a second-generation didemnin isolated from the Mediterranean tunicate Aplidium albicans, which inhibits protein and DNA synthesis and induces apoptosis and a G1 cell cycle arrest in cancer cells independently of p53 and MDR expression. In a phase I study, aplidine was administered as a 24-hour intravenous infusion every 2 weeks to patients with advanced cancers, including 6 with MTC [149]; minor responses and prolonged tumour stabilization (>6 months in 4 patients) were observed in patients with MTC.
Ongoing Clinical Trials with Novel Agents
Because RET is a growth fractor receptor with limited expression, and there are both germline and somatic mutations, it has been an attractive candidate for targeted therapy. There has been significant experimental evidence showing that RET inhibition leads to growth inhibition and apoptosis in MTC cells [150]: as a result, different strategies have been developed to inhibit the kinase function of RET. Loss of heterozygosity in the von-Hippel-Lindau (VHL) disease tumour suppressor locus occurs at the somatic level in MEN 2 patients [151]. Because loss of VHL protein leads to increased expression of hypoxia-inducible transcription factor, thereby promoting expression of VEGF and tumour angiogenesis, anti-angiogenic therapy is also a good candidate for MTC therapy.
There are a number of RET kinase inhibitors which share the property of binding to the RET ATP-binding pocket; these include vandetanib, sorafenib, sunitinib, imatinib, pazopanib, axitinib, motesanib, gefitinib and XL 184 [150]. Many of these small molecules are not only RET kinase inhibitors but multikinase inhibitors effective on VEGF-R1, VEGF-R2 and VEGF-R3, PDGF-Rβ and EGF-R, with varying affinities, and thus often affecting multiple signalling pathways. The results of most of the phase II trials that have focused on the efficacy of these drugs on MTC have not as yet been published, except in abstract form at scientific meetings, as shown below. There are also a number of ongoing open clinical trials with some of these agents (table 1).
Vandetanib is an oral, small-molecule tyrosine kinase inhibitor which inhibits RET, VEGF-R2, VEGF-R3 and, at higher concentrations, EGF-R [152]. Vandetanib inhibits most of the activated forms of RET, with the exception of RET molecules with mutations in codon 804. On the basis of preclinical evidence, a multicentre, open-label phase II trial of vandetanib was performed in locally advanced, unresectable or metastatic hereditary MTC patients with germline RET mutations [153]: 30 patients were enrolled and 300 mg/day vandetanib treatment was given. The primary objective of the study was to assess objective tumour response by RECIST criteria every 3 months. Based on site investigator assessments, 20% of patients experienced a partial response, and 30% showed stable disease for at least 24 weeks. Calcitonin levels showed a 50% decrease from baseline in almost two-thirds of patients, and this was maintained for at least 6 weeks. Adverse events occurring in >50% of patients were rash (73%), diarrhoea (67%), fatigue (57%) and nausea (53%). In another phase II open-label study, 19 patients with locally advanced or metastatic hereditary MTC were enrolled to assess 100 mg/day vandetanib therapy [154]. On disease progression, eligible patients were planned to enter post-progression treatment with vandetanib 300 mg until a withdrawal criterion was met. Preliminary objective tumour assessments demonstrated partial responses in 2 patients, stable disease >24 weeks in 6 patients and progressive disease in 2 patients, yielding an objective response rate of 10.5% (2/19) and a disease control rate of 42.1% (8/19). This study showed that vandetanib 100 mg/day could also be effective in MTC. An international phase I/II study of vandetanib is currently recruiting patients (table 1). An international, phase II, randomized, double-blinded, placebo-controlled, multicentre study to assess the efficacy of vandetanib versus placebo in subjects with unresectable, locally advanced or metastatic MTC, and a phase II, open-label study to assess the efficacy and tolerability of vandetanib 100 mg monotherapy in subjects with locally advanced or metastatic hereditary MTC, have completed recruitment of subjects and the studies are ongoing [155].
Sorafenib is an oral, biaryl urea compound that targets RET, VEGF-R2, VEGF-R3, PDGF-Rβ, FLT-3, KIT and FGF-R1, as well as the serine-threonine kinase B-RAF and p38 MAPK [156]. Its anti-RET activity in particular has made it a potential therapeutic agent for MTC. Following a small pilot study including 5 subjects with metastatic MTC [157], an open-label phase II study was conducted in advanced/metastatic MTC (10 patients) as well as in patients with differentiated thyroid cancer (DTC) [158]; 10 patients had follow-up at 3 months and 2 of these patients had follow-up at 6 months. The remainder have been on the drug for <3 months and could not as yet be assessed. At 3 months, the best objective overall response for 9 patients has been stable disease, although 1 patient has demonstrated a partial response. At 6 months 2/2 patients have stable disease. Biochemically, 7 out of 7 assessable patients have demonstrated a partial response. Common adverse events include diarrhoea (grade 1/2 = 50%), hand-foot syndrome (grade 1/2 = 39%, grade 3 = 17%), other skin toxicity (grade 1/2 = 44%, grade 3 = 6%), alopecia (grade 1/2 = 28%), hypertension (grade 1/2 = 22%, grade 3 = 6%), infection (grade 1/2 = 17%, grade 3 = 6%) and nausea (grade 1/2 = 17%). Currently, 2 phase II trials are recruiting advanced MTC patients for sorafenib treatment (table 1). Based on the intimate connection of Ras and Raf kinases, it was hypothesized that inhibiting both Ras (upstream) with a farnesyltransferase inhibitor (tipifarnib) and B-Raf (downstream) with a Ras inhibitor (sorafenib) may confer synergistic antitumour effects. In a sporadic MTC case with advanced, progressive disease Hong et al. [159] reported that the combination of sorafenib and tipifarnib slowed disease progression. A phase I study has been designed to include subjects with MTC treated with sorafenib [160].
Sunitinib is another oral, tyrosine kinase inhibitor drug which targets VEGF-R1, -2, -3, RET, KIT, PDGF-Rα, PDGF-Rβ, FLT3 and colony-stimulating factor receptor type 1 (CSF-1R) [16, 162]. Kelleher et al. [163]have reported a metastatic, progressive MTC case treated with sunitinib, 50 mg/day for 28 days, followed by 14 days of no treatment. After 2 cycles of therapy, the patient’s longstanding diarrhoea had resolved and a clinically enlarged right cervical lymph node had become impalpable. In a phase II study conducted in patients with refractory DTC cancer (n = 37) and MTC (n = 6), 6-week cycles of sunitinib maleate 50 mg q.d. on a 4-week on/2-week off schedule was given [164]. The primary endpoints were RECIST and biochemical response rate.The best response in MTC patients was stable disease in 83%, with progressive disease in 17%. The most common drug-related adverse events and incidence included fatigue (79%), diarrhoea (56%), palmar-plantar erythrodysesthesia (53%), neutropenia (49%), and hypertension (42%). Grade 3/4 toxicity included neutropenia (26%), thrombocytopenia (16%), hypertension (16%), fatigue (14%), palmar-plantar erythrodysesthesia (14%), and gastrointestinal tract events (14%, 2 diarrhoea, 1 tracheo-oesophageal fistula, 1 gastric haemorrhage, 1 gastric ulcer, and 1 gastritis). Currently, there are 3 trials recruiting MTC patients for sunitinib treatment (table 1).
Axitinib is an oral, small-molecule tyrosine kinase inhibitor that inhibits PDGF-Rβ and c-KIT and also effectively blocks VEGF-R1, -2, -3 at subnanomolar concentrations but with no known anti-RET activity [162]. It has been tested in a multicentre, open-label phase II study in patients with advanced or metastatic thyroid cancer including MTC starting at a dose of 5 mg twice daily [165]. In the 12 MTC patients included in the study, stable disease was noted in 42%. Common adverse events include fatigue, stomatitis, proteinuria, diarrhoea, hypertension and nausea.
Imatinib inhibits the product of KIT proto-oncogene and PDGF-Rβ, as well as RET (but only at high concentrations [162]). A phase II study was initiated using 600 mg imatinib daily with a possible dose increase to 800 mg in case of progression [166]: 15 patients with disseminated MTC were treated for up to 12 months, but no objective responses were observed and imatinib treatment induced considerable toxicity in patients with MTC. Imatinib treatment was used in a multicentre phase II study in patients with solid tumours [167]. The patient group included 6 subjects with MTC. Imatinib was initiated at a dose of 400 mg/day, with possible dose escalation within 1 week to 600 mg/day and an option to raise the dose to 800 mg/day in the event of progression and in the absence of safety concerns for a period of up to 12 months. No objective responses were observed. Disease progression occurred in 4 patients, and treatment was discontinued in 2 patients due to adverse events.In another open-label clinical trial, 9 patients (8 with sporadic and 1 with hereditary MTC) with unresectable, measurable, progressive metastases were treated with imatinib mesylate 600 mg daily [168]. In this study, stable disease occurred in 5 patients for up to 6 months and in 1 patient for up to 12 months, with a median duration of progression-free survival of 6 months; 4 patients had progressive disease after 12 months. Thus, although in this study imatinib was well tolerated, in general no tumour remission was observed and only transient stable disease was achieved in occasional patients.
Gefitinib, is an EGF-R inhibitor and has been used at a dose of 250 mg/day in a phase II open-label trial in 27 patients with radioiodine-refractory, locally advanced, or metastatic mixed cohort of thyroid cancer [169]. With the overall response rate being the primary endpoint, all 4 of the MTC patients included in the study had progressive disease within 3 months of enrollment and serum calcitonin and CEA levels, which were elevated at baseline, rose progressively throughout the treatment.
Motesanib diphosphate is an oral small-molecule tyrosine kinase inhibitor targeting VEGF-R1, -2, -3, RET, PDGF-Rβ and KIT [162]. On the basis of in vitro studies and a small phase I study, a multicentre, open-label phase II trial including 91 progressive or symptomatic, either sporadic- or hereditary-MTC patients was conducted. With a median follow-up of 32 weeks, the objective tumour response was 1%, stable disease 80% (durable stable disease ≥weeks, 24%) and progressive disease = 10% [170]. The drug was well tolerated, except for side effects such as fatigue, nausea, diarrhoea and hypertension. An unexpected side effect was an average increase of 30% in the doses of levothyroxine to maintain euthyroidism.
XL 184 is a VEGF-R2, hepatocyte growth factor receptor (c-MET), c-KIT, FLT-3, endothelial cell RTK receptor (Tie-2), RET and PDGF-Rβ inhibitor [162]. In a phase I study carried out in 55 patients with advanced malignancies, including 13 patients with MTC, XL 184 was tested at several dose levels [171]. All evaluated MTC patients had reductions in plasma calcitonin and CEA levels. Of the 10 patients who were evaluated, 7 had stable disease, 3 had a partial response and XL 184 was concluded to show antitumour activity in MTC patients. In another phase I study, XL 184 was given to 22 MTC patients [172]. Eight of the 16 MTC patients with measurable disease had a partial response (50%, 5 confirmed) with all others experiencing prolonged stable disease; overall disease control rate (progressive disease + stable disease >3 months) is currently 100%. Three MTC patients had non-measurable disease and 3 are too early to evaluate. Three partial responses were reported after only 4 weeks of daily XL184. Most patients have had substantial reductions in plasma calcitonin and CEA. A study is currently recruiting MTC patients for testing XL 184 (table 1).
Another new drug being tested in MTC is thalidomide, which is known to inhibit VEGF and basic fibroblast growth factor (bFGF)-mediated angiogenesis [173]. A phase II trial for treatment of patients (n = 36) with radioiodine-unresponsive and rapidly progressive thyroid cancer was designed [173]. Daily thalidomide treatment was started at 200 mg, increasing over 6 weeks to 800 mg or maximum tolerated dose. Toxicities were dose limiting in the majority of the subjects, and the most common adverse events included somnolence, peripheral neuropathy, constipation, dizziness and infection. Of the 7 MTC patients included in the study, 5 were eventually able to be evaluated, but only 1 was reported to have stable disease or a partial response; however, dose and treatment period regarding this patient were not given in detail.
In a phase I trial of the bombesin/gastrin-releasing peptide (BN/GRP) antagonist RC3095 in patients with advanced solid malignancies, 1 patient with progressive MTC was also included [174]. BN/GRPs are shown to bind selectively to cell surface receptors, stimulating the growth of various types of malignancies in murine and human models, and the novel BN/GRP synthetic receptor antagonist, RC-3095, was able to produce long-lasting tumour regressions in murine and human tumour models in vitro and in vivo. RC-3095 was given once or twice daily at doses ranging from 8 to 96 µg/kg. A short-lasting minor tumour response was observed in the patient with GRP-expressing progressive MTC. Due to the occurrence of local toxicity at the injection site, the dose escalation procedure could not be fully evaluated up to a maximum tolerated dose. Thus, a recommended dose of RC-3095 for phase II trials could not be clearly established.
The effects of a 12-week course of somatostatin analogue therapy on calcitonin and CEA in patients with advanced MTC were evaluated in a study of 5 patients (2 men and 3 women, aged 35–57 years) [175]. One patient was treated with subcutaneous octreotide (100 µg/8 h), 3 patients received slow-release lanreotide (30 mg/14 days i.m.), and a further one received octreotide LAR (30 mg/28 days i.m.). The therapy was well tolerated, but changes in hormonal levels were variable and inconsistent. In conclusion, therapy with different formulations of octreotide and lanreotide did not seem to modify serum concentrations of calcitonin and CEA in patients with recurrent MTC.
Although most of the tyrosine kinase inhibitors lack selectivity for RET, adverse effects seem manageable and these drugs may indeed find a place in the treatment of RET-associated cancer. However, it should be kept in mind that not all mutations are sensitive to these drugs. In a study by Carlomagno et al. [176], it was demonstrated that most oncogenic MEN-2-associated RET kinase mutants are highly susceptible to PP1, PP2 and vandetanib inhibition. In contrast, MEN-2-associated exchange of bulky hydrophobic leucine or methionine residues for valine 804 in the RET kinase domain caused resistance to the 3 compounds. Replacement of valine 804 with the small-amino-acid glycine renders the RET kinase even more susceptible to inhibition than the wild-type kinase. These data identify valine 804 of RET as a structural determinant mediating resistance to pyrazolopyrimidines and 4-anilinoquinazolines. In a further study, the same group demonstrated that sorafenib inhibited a codon 804 mutation at higher doses compared to wild-type RET [177].
In the phase II trials discussed above, RECIST guidelines were used for patient evaluation, other than in the thalidomide study. This point is important in terms of bringing standardization to interpretation of treatment responses. However, tumour responses were observed in only a fraction of patients, which suggests that even multikinase inhibition including RET may not be enough to treat all MTC tumours. However, RECIST criteria may not always be appropriate in slow-growing tumours which may regress by central necrosis, which might not necessarily be demonstrable on simple measurement of lesion diameters.
Conclusions
MTC is still one of the most challenging cancers for both physicians and patients. We now understand a considerable amount regarding the molecular biology of MTC, yet the major effective treatment modality remains surgery. Although many drugs are being tested, and many are under investigation, there is still no universally recommended drug treatment regimen for residual disease and recurrences. While tyrosine kinase inhibitors, particularly those affecting RET activity such as vandetanib, sorafenib and sunitinib, are promising, the low rate of partial responses and absence of complete responses in any of the various trials of monotherapy emphasize the need for new and more effective single agents or combinations of therapeutic agents with acceptable toxicity. The use of RET inhibitors may render these tumours more sensitive to chemotherapy, but as yet no major combination trials have been published. Nevertheless, this is an exciting time to explore the therapy of these tumours, and as research into their pathogenesis continues it is likely that more effective treatments will become available.
Acknowledgements
M.C. is currently at Selcuk University, Meram School of Medicine, Division of Endocrinology and Metabolism, Konya, Turkey, and was awarded a grant by The Society of Endocrinology and Metabolism of Turkey to study at the Centre for Endocrinology, Barts and The London School of Medicine and Dentistry, London.
