

Abstract
Background: This paper examines the current status of research on the efficacy and effectiveness of antidepressants. Methods: This paper reviews four meta-analyses of efficacy trials submitted to America’s Food and Drug Administration (FDA) and analyzes STAR*D (Sequenced Treatment Alternatives to Relieve Depression), the largest antidepressant effectiveness trial ever conducted. Results: Meta-analyses of FDA trials suggest that antidepressants are only marginally efficacious compared to placebos and document profound publication bias that inflates their apparent efficacy. These meta-analyses also document a second form of bias in which researchers fail to report the negative results for the pre-specified primary outcome measure submitted to the FDA, while highlighting in published studies positive results from a secondary or even a new measure as though it was their primary measure of interest. The STAR*D analysis found that the effectiveness of antidepressant therapies was probably even lower than the modest one reported by the study authors with an apparent progressively increasing dropout rate across each study phase. Conclusions: The reviewed findings argue for a reappraisal of the current recommended standard of care of depression.
Introduction
When medications are evaluated to determine their applicability to evidence-based clinical practice, it is important to assess their efficacy in randomized, double-blind, placebo-controlled trials (RCT) in addition to determining their effectiveness in treating real-world patients under conditions that simulate real-world practice.
Efficacy
Due to long-held concerns about publication bias inflating perceived efficacy [1,2,3,4] and the resulting adverse impact on evidence-based care, public-minded researchers have long argued for a comprehensive registration data repository providing full access to drug trial protocols and results [1,5,6]. Though by no means complete, America’s Food and Drug Administration (FDA) maintains a large repository of RCT trials as part of its new drug application process. Prior to conducting new drug application trials, drug companies must register them with the FDA, which includes pre-specifying the primary and secondary outcome measures and means of analysis. Pre-specification is essential to ensure the integrity of a trial and enables the discovery of when investigators selectively publish the measures that show the outcome the sponsors prefer following data collection and analysis, a form of researcher bias known as HARKing [7] or ‘hypothesizing after the results are known’.
Rising et al. [8] recently published a meta-analysis of all efficacy trials for new drugs approved by the FDA from 2001 to 2002 and the subsequent publication status of these trials 5 years later. Key findings were:
• New drug application studies with favorable outcomes were almost five times more likely to be published as those with unfavorable ones.
• 26.5% of pre-specified primary outcome measures were omitted from journal articles of new drug trials.
• Of the 43 primary measures not supporting efficacy, 20 (47%) were not included in the published results.
• 17 measures were only presented in the published studies and 15 of these showed positive effects for the new drug.
The analysis of Rising et al. [8] documents significant publication bias that inflates the apparent efficacy of new drugs. In addition to selective publication of positive trials, more disturbingly researchers at times fail to report the negative results of pre-specified primary outcome measures while highlighting in published studies positive results from a secondary or even a new measure as though it was their primary measure of interest. Besides casting doubt on the accurate reporting of individual drug trials in journal articles, their findings also directly challenge the validity of meta-analyses covering specific drugs and drug classes when limited to published studies.
Several antidepressant meta-analyses have been conducted using the FDA data repository to avoid the inflationary effects of publication bias. In 2008, Turner et al. [9] reviewed 74 trials of 12 antidepressants to assess subsequent publication bias and its influence on apparent efficacy.
In their meta-analysis, antidepressant studies with favorable outcomes were 16 times more likely to be published as those with unfavorable ones. According to the FDA scientific reviews though, only 38 trials (51%) found positive drug/placebo differences and 37 were subsequently published. The FDA judged the remaining 36 studies to be either negative (24 studies) or questionable (12 studies) – that is, no difference on the primary outcome but significant findings on a secondary measure. Only 3 (8%) were published reporting negative results, while the remaining 33 were either not published (22 studies) or published as though they were positive (11 studies) in contradiction to the FDA conclusions. Similar to Rising et al. [8], Turner et al. [9] report that in these 11 studies the authors did not report their negative results for the pre-specified primary outcome measure and instead highlighted positive results from a different measure as though it was their primary measure of interest.
Turner et al. [9] next compared the effect size derived from the FDA repository to that from the 51 published studies. This analysis found that the weighted mean effect size in the FDA data set was a modest 0.31 (95% confidence interval, 0.27–0.35) versus 0.41 (95% confidence interval, 0.36–0.45) in the published studies indicating a 32% inflation of the apparent efficacy of antidepressants. Their findings are similar to those of Kirsch et al. [10] in 2002 in a meta-analysis of 47 trials of 6 FDA-approved antidepressants. Though statistically significant due to the large combined number (n = 6,944), they found that the weighted mean difference between groups on the 17-item Hamilton Rating Scale for Depression (HRSD) was only 1.8 points and 57% of the trials found no significant drug/placebo differences [11].
Kirsch et al. [10] and Kirsch and Antonuccio [12] also examined patients’ response as a function of dosing strength and time. These analyses found no advantage for higher antidepressant dosing. Regarding the common belief that drug effects are more enduring than placebo effects, they found that while patients’ initial positive responses to both decrease over time, the correlation was higher for antidepressants (r = –0.84) than placebos (r = –0.62), suggesting that the effects of antidepressants diminish more rapidly than those of placebo.
In 2008, Kirsch et al. [13] examined the relationship between depression severity and efficacy in all 35 trials (n = 5,133) of four new-generation antidepressants. This analysis found no clinically significant difference (defined as a drug/placebo difference of ≥3 on the HRSD) between antidepressants and placebos as a function of severity except for the most severely depressed patients (i.e. those with a ≥29 HRSD score). However, even this difference was due to a decreased placebo response in more severely depressed patients, not an increased response to antidepressants.
Also in 2008, Barbui et al. [14] analyzed 40 paroxetine studies (29 published/11 unpublished; n = 6,391) using early trial termination for any reason (i.e. dropout) as the primary outcome assessing it as the best available ‘hard measure of treatment effectiveness and acceptability’ [[14], p. 296]. Their analysis found no drug/placebo difference on this measure in contrast to a secondary one that paroxetine was clinically superior to placebo in patients’ likelihood of achieving a ≥50% reduction in depressive symptoms. They also found that significantly more paroxetine patients dropped out due to side effects and increased suicidal tendencies. In a subsequent analysis though of these same studies, the apparent clinical superiority of paroxetine disappeared after statistically controlling for the differences in drug/placebo side effects [[15], cited in [16], p. 19]. This finding is similar to that of Greenberg et al. [17] of an exceptionally high correlation between side effects and improvement in fluoxetine/placebo trials and suggests that when it does occur, the apparent superiority of antidepressants may be due to unblinding among study patients (and raters) given the greater frequency of side effects in active drug groups and patients’ education about said effects as part of informed consent [18,19]. Kirsch [[16], pp 7–22] and Kirsch and Sapirstein [20], among others [18,19,21], argue that side effects enhance the placebo effects of antidepressants by confirming to patients that they are taking the active medication and thereby increasing their expectation of improvement. Given the often small drug/placebo differences in these studies, it would not take much such unblinding to account for positive results when they do occur. On the other hand, the fact that many RCTs fail despite significant drug/placebo side effect differences suggests that said effects alone are not sufficient to consistently result in greater improvement even if they do contribute to unblinding.
The analyses performed by Rising et al. [8] and Turner et al. [9 ]document that readers should be wary when researchers replace their pre-specified primary outcome measure with a new one. This concern is reinforced by recent analyses documenting widespread selective outcome reporting in industry-sponsored research and the inflationary effect that often occurs when the pre-specified primary outcome measure is not used to report findings [22,23]. Perhaps the most troubling implications of the Rising-Turner-Kirsch-Barbui findings are that journal readers, seeking articles to guide evidence-based practice, may have been misled by meta-analyses and reviews based on biased published articles on antidepressant efficacy.
However, some explain only marginal superiority of antidepressants by the fact that subjects enrolled in RCTs do not necessarily present with adequate illness severity. Lieberman et al. [24] observed that early RCTs often enrolled hospitalized patients who were less responsive to placebo, whereas more recent trials typically enroll highly selected outpatients, contacted through mass media advertisements, who may be less severely depressed. In a meta-analysis of 75 RCTs published between 1981 and 2000, Walsh et al. [25] showed that the response to both antidepressants and placebos has increased over time with a significant positive correlation between year of publication and response. Parker [26] argues that this progressively increasing response to both compromises the ability to differentiate truly efficacious antidepressants from placebos, particularly among less severe patients.
Fava et al. [27] notes that the analysis of Walsh et al. [25] likely understates placebo response rates since most failed trials go unpublished; they estimate that the true rate is 35–45%. They then explore various potential causes for failed trial findings (e.g. measurement errors or diagnostic heterogeneity) and propose a new study design that might reduce placebo rates and thereby the number required to differentiate efficacious antidepressants from placebos. Both Fava et al. [27] and Otto and Nierenberg [28] observe that only two RCTs showing drug superiority are required for FDA approval regardless of how many were conducted, and both cite the example of paroxetine that took nine trials to get the two necessary to ‘win’ approval [29]. The apparent lack of significant adverse consequences to drug companies from failed trials (other than added costs and delayed time to market) may have fostered a production-oriented mind-set favoring trial quantity over quality (since it takes only two to win, and losses are not counted) too often resulting in flawed science and thereby the ensuing vigorous debate over methodology and interpretation while furthering the disconnect between trial findings and their application to clinical practice.
In an analysis of psychiatric outpatients with major depressive disorder (MDD), Zimmerman et al. [30] found that RCTs would have excluded 86% due to their having a comorbid anxiety or substance use disorder, insufficient depressive symptoms, and/or current suicidal ideation. Similarly, a post hoc analysis of STAR*D (Sequenced Treatment Alternatives to Relieve Depression) patients, the largest antidepressant effectiveness study ever conducted, found that 77.8% would have been excluded from RCTs due to having a baseline HRSD score ≤19, more than one concurrent medical condition, more than one comorbid psychiatric disorder, and/or a current depressive episode lasting >2 years [31]. This analysis found that STAR*D patients who met RCT inclusion criteria had greater likelihood of remission than those more representative of the vast majority seeking care (34.4 vs. 24.7% remission rate). As Wisniewski et al. [31] note, by enrolling more representative patients RCT results would better estimate the benefit of an antidepressant in practice and may also reduce placebo response rates and the associated risk of failed trials. Likewise, Parker [[26], p. 2] argues that the apparent limited efficacy of antidepressants may not be related to the modest effects of these compounds but rather to RCT design and methodological issues, ‘whereby the ‘‘apples’’ assessed in such studies do not correspond to the ‘‘oranges’’ of clinical practice’.
While the relative efficacy of antidepressants is not settled, progress will only come as RCTs enroll representative MDD samples for which the medication is intended under conditions that simulate real-world practice. Such trials should follow Gaudiano and Herbert’s [19] recommendations for distinguishing between specific and nonspecific treatment effects, the first one being having an active placebo arm to reduce the likelihood of unblinding and thereby control the potential role of side effects in enhancing patients’ expectation of improvement.
Real-World Effectiveness of Antidepressants
In conformity with this goal of enrolling more representative MDD patients, the NIMH-funded STAR*D study [32,33,34,35,36,37,38]:
• enrolled 4,041 real patients seeking care versus persons responding to advertisements for depressed subjects;
• included patients with comorbid medical and psychiatric conditions as well as those whose current MDD episode was >2 years;
• included patients currently undergoing antidepressant treatment provided that there was not a history of nonresponse or intolerance to either step-1 or step-2 protocol antidepressants;
• used ‘remission’ versus ‘response’ as the primary criterion of successful treatment which is a higher clinical standard than used in prior effectiveness studies;
• provided 12 months of continuing care while monitoring the durability of treatment effects versus only reporting acute-care improvement.
STAR*D provided a very high quality of free acute and continuing care to maximize the likelihood that MDD patients would achieve remission and maintain it. Table 1 describes in detail the high quality of treatment and the extensive efforts of STAR*D to retain patients. The treatment protocol included state-of-the-art acute care to achieve remission followed by 1 year of continuing care for all patients who achieved a satisfactory clinical response. The goal of continuing care was to maintain remission and prevent relapse [[39], p. 15].
Highest quality of acute and continuing care to maximize remissions while minimizing relapse and dropouts

The continuing-care phase of STAR*D also provided a real-world test of the practice guideline of the American Psychiatric Association (APA) that ‘following remission, patients who have been treated with antidepressant medications in the acute phase should be maintained on these agents to prevent relapse’ [[40], p. 15]. This guideline received the highest confidence rating of the expert panel.
STAR*D was designed to provide guidance in selecting the best ‘next-step’ treatment for the many patients who fail to get adequate relief from their initial selective serotonin reuptake inhibitors (SSRI) trial by evaluating the relative efficacy of eleven pharmacologically distinct treatments [41]. Cognitive therapy (CT) was also an option in step 2, but too few patients included it as an acceptable treatment resulting in only 101 contributing data after randomization [33]. CT was therefore excluded from the primary step-2 switch and augmentation articles [33,34]. Possible reasons so few patients found CT acceptable included: (1) biased self-selection since all patients started on citalopram in step 1; (2) the added costs of CT which STAR*D did not cover whereas it covered all medication and physician visit costs, and (3) CT patients had to go to another site to see a new non-physician professional [[42], pp 748–749]. Despite these impediments, in a subsequent article STAR*D reported that the 101 step-2 patients who received CT ‘(either alone or in combination with citalopram) had similar response and remission rates to those assigned to medication strategies’ [[42], p. 739].
The conclusion section of the research design article of STAR*D states: ‘STAR*D uses a randomized, controlled design to evaluate both the theoretical principles and clinical beliefs that currently guide the management of treatment-resistant depression in terms of symptoms, function, satisfaction, side-effect burden, and health care utilization and cost estimates. Given the dearth of controlled data, results should have substantial public health and scientific significance, since they are obtained in representative participant groups/settings, using clinical management tools that can easily be applied in daily practice’ [[43], p. 136].
Given the ‘substantial public health and scientific significance’ of STAR*D in evaluating the effectiveness of antidepressants when optimally delivered to real-world patients, it is critical that the methodology and findings of STAR*D are presented accurately.
Change in One of the Outcome Measures
As designed, the HRSD was the pre-specified primary measure of STAR*D and the Inventory of Depressive Symptomatology-Clinician-Rated (IDS-C30) the secondary one for identifying ‘remitted’ (i.e. those with a score ≤7 HRSD) and ‘responder’ (i.e. those with a ≥50% reduction in depressive symptoms) patients [[41], p. 476, [43], p. 123]. These measures were obtained by research outcome assessors (ROAs) blind to treatment assignment at entry into and exit from each treatment level, and every 3 months during continuing care. Additional measures assessing symptoms, level of functioning, satisfaction, quality of life, side effect burden, and health care utilization were obtained by an interactive voice response (IVR) telephone system on the same schedule as the HRSD and IDS-C30 [[43], p. 129] (table 2).
As mentioned earlier, STAR*D was designed to identify the best next-step treatment for the many patients who fail to get adequate relief from their initial SSRI trial. Due to the high study dropout rate in STAR*D which frequently resulted in missing exit IDS-C30 and HRSD assessments (and thereby the required imputation level made these assessments relatively uninformative), the statistical analytical plan was revised with input from the Data Safety and Monitoring Board prior to data lock and unblinding.
The revised statistical analytical plan of STAR*D dropped the IDS-C30 and replaced it with the Quick Inventory of Depressive Symptomatology-Self-Report (QIDS-SR), a tool developed by the principal investigators of STAR*D that is highly correlated with the HRSD (the primary measure) since it was administered at every visit [44,45,46]. In the step-1 to step-4 studies, the QIDS-SR was the secondary measure for identifying remissions and sole measure for identifying responders while the HRSD was the primary measure for identifying remitted patients [32,33,34,35,36,37].
As originally planned, STAR*D used the QIDS-SR in two ways. The research version was IVR administered on the same schedule as the other measures during steps 1–4 and as an ‘interim’ monthly measure during continuing care [[39], p. 120, table 3].
Patients also completed a paper-and-pencil QIDS-SR at the beginning of each clinic visit along with two self-rated side effect measures and a medication adherence questionnaire. These four self-assessments were overseen by non-blinded clinical research coordinators who reviewed the results to make certain that all items were completed and then saw the patient to administer the QIDS-C (the clinician-administered version with the identical 16 questions and response options as the QIDS-SR), the Clinical Global Impression Improvement Scale, and discuss with the patient any symptoms and side effects that he/she was experiencing as well as present patient education materials [[39], p. 75].
The clinical research coordinators then recorded the six measures on the clinical record form for the treating physician’s review before he/she saw the patient ‘to provide consistent information to the clinicians who use this information in the protocol’ [[43], p. 128]. In this way, the paper-and-pencil QIDS-SR was one of the ‘clinical management tools that can easily be applied in daily practice’ [[43], p. 136] of STAR*D and used as such in the ‘measurement-based’ system of care of STAR*D as STAR*D states: ‘To enhance the quality and consistency of care, physicians used the clinical decision support system that relied on the measurement of symptoms (QIDS-C and QIDS-SR), side-effects (ratings of frequency, intensity, and burden), medication adherence (self-report), and clinical judgment based on patient progress’ [[32], p. 30]. This distinction between the originally intended use of the QIDS-SR as a clinical tool versus research measure is made explicit in both tables of the design article (tables 2, 3) [[43], pp 128–129] and the Clinical Procedure Manual of STAR*D (tables 2, 4) [[39], pp 119, 121].


The revised statistical analytical plan did not change any of the next-step comparison results of STAR*D, as both the QIDS-SR and HRSD found no significant group differences between treatments in all five comparisons. However, this decision appears to have inflated the reported remission and response rates of STAR*D. As stated in the step-1 article: ‘Higher remission rates were found with the QIDS-SR than with the HRSD because our primary analyses classified patients with missing exit HRSD as nonremitters a priori. Of the 690 patients with missing exit HRSD scores, 152 (22.1%) achieved QIDS-SR remission at the last treatment visit’ [[32], p. 34]. In the six step-1 to step-4 studies, there were 1,192 HRSD-identified remissions versus 1,398 QIDS-SR ones, an increase of 17.3% (table 4), and the major summary article of STAR*D only used the QIDS-SR to report its step-by-step acute and continuing-care findings [38].
Change in Eligibility for Analysis Criteria
The step-1 article of STAR*D states that eligible patients ‘had a non-psychotic major depressive disorder determined by a baseline HRSD score≥14’ [[32], p. 29]. This is a modest symptom severity threshold (see Davidson et al. [47] with a ≥20 HRSD inclusion threshold for example) though similar to many MDD patients seeking treatment [30].
Of the 4,790 patients administered the screening HRSD (completion time: 15 min [[39], p. 118]), 4,041 were started on citalopram in their baseline visit [[39], p. 17, [48]]. Of these patients, 3,110 scored ≥14 on the more thorough and blinded ROA-administered HRSD (completion time: 20–25 min [[39], p. 118]), 234 of whom failed to return for a follow-up visit. For the resulting 2,876 step-1 patients eligible for analysis [[32], fig. 1], their baseline HRSD scores averaged 21.8 [32,]table 1, row 2]. There were also 607 patients reportedly excluded (but later included – see discussion below) because their score <14 signified only mild depression and 324 patients were reportedly excluded (but later included) because they lacked a baseline ROA-administered HRSD [[32], fig. 1].
The subsequent step-2 to step-4 articles of STAR*D continued to state that all patients had ‘non-psychotic major depressive disorder’ and did not clarify a deviation from eligibility of step 1 for analysis criteria [32,33,34,35,36,37]. Specifically, the 607 patients who scored <14 in their baseline ROA assessment along with the 324 patients with no such assessment received citalopram in step 1, progressed to subsequent acute and continuing-care treatments, and were included in step-2 to step-4 articles and also in the summary article. Thus, 931 of the 4,041 STAR*D patients (23%) did not have a ROA-administered HRSD ≥14 when enrolled into the study.
The effect of including these patients’ data was to lower the average step-1 HRSD from 21.8 to 19.9 [[38], table 2, row 2]. STAR*D also included these 931 patients to recalculate the step-1 remission rate of citalopram in its summary article. In so doing, the step-1 remission rate was inflated to 36.8% from the original 32.8% of the step-1 article both based on the lenient QIDS-SR determination.
Perhaps a more serious problem is trying to determine the effect of these 931 patients in changing the relapse rate during continuing care. Initial symptom severity is a powerful predictor of relapse – with less depressed patients far less likely to relapse. For patients who entered the study with a HRSD <14, there is a special conundrum because STAR*D defined relapse as a HRSD ≥14. This means that 607 patients, in order to be counted as relapsed, had to score worse during continuing care than when they first entered the study.
In addition, a failure to consider dropout permitted STAR*D to assert a 67% ‘cumulative’ remission rate after up to four medication steps [[38], p. 1910]. STAR*D authors arrived at this figure simply by adding together its inflated QIDS-SR remissions in steps 1–4. STAR*D acknowledges that this assertion assumes no dropouts and the same remission rate for persisting patients as those who exited. These assumptions though are not true in the real world and were certainly not true in STAR*D since more patients dropped out from the study in each step than had a remission (table 3). Furthermore, comparing persisting patients’ remission rates to dropouts is not possible since dropouts do not provide data.
Table 3 presents the HRSD-determined remission and dropout rates of STAR*D for steps 1–4. These data are quite important for a clinical understanding of the effectiveness of antidepressants in real-world patients receiving ‘measurement-based’ state-of-the-art treatment:
• in step 1, 25.4% of patients had a remission while 26.6% dropped out [49];
• in step 2, 25.1% of patients had a remission while 30.1% dropped out;
• in step 3, 17.8% of patients had a remission while 44.8% dropped out;
• in step 4, 10.1% of patients had a remission while 60.1% dropped out.
Continuing-Care Findings
One of the most important questions in evaluating antidepressants is how durable are the remissions. A major STAR*D contribution was to provide 12 months of follow-up data on remitted and improved patients’ continued treatment. Patients who achieved remission during acute care were strongly encouraged to enter continuing care. In addition, responder patients who failed to attain remission and did not want to continue to the next acute-care step were encouraged to enter continuing care.
The protocol of continuing care ‘strongly recommended that participants continue the previously effective acute treatment medication(s) at the doses used in acute treatment’ [[38], p. 1908]. This recommendation is consistent with the APA continuation phase guideline that ‘following remission, patients who have been treated with antidepressant medications in the acute phase should be maintained on these agents to prevent relapse’ [[40], p. 15]. However, the STAR*D protocol was more naturalistic than the APA guideline in that physicians were allowed to make ‘any psychotherapy, medication, or medication dose change’ [[38], p. 1908] they deemed necessary to maximize patients’ likelihood of sustaining remission. This included scheduling additional visits if depressive symptoms returned and/or intolerable side effects emerged [[39], p. 78].
STAR*D made strong efforts to maximize retention and collect follow-up data. These efforts included continued use of all acute-care patient retention strategies during continuing care, prompting patients prior to all research outcome assessment phone calls, and paying patients USD 25.00 for taking said assessments as well as permitting patients to remain in the study if they moved from the area (table 1). Furthermore, informed consent was re-obtained for all patients entering continuing care to ensure their understanding of its treatment and outcome assessment procedures and expectations [[39], p. 68].
For evaluating the outcome of continuing care, STAR*D authors decided to use the IVR-administered QIDS-SR that was originally intended as only an interim monthly measure during continuing care [[39], p. 120, table 3], not the pre-specified HRSD and IDS-C30. If the patient called in and scored ≥11 on the QIDS-SR, relapse was declared; that score (said to correspond to a HRSD ≥14) indicated moderate-to-more-severe depression. Given this, it is important to note that STAR*D was not reporting the rate that remitted patients sustained remission, only the rate at which they relapsed while remaining in continuing care.
In calculating relapse, STAR*D authors decided not to use intent-to-treat procedures in which dropouts would count as continuing-care failures (despite the separate informed consent process) but instead use the data from patients who called into the IVR system once (or more) during the 12 months. The calculated relapse rate was the proportion of patients relapsing of those who made at least one such call and reported a ≥11 QIDS-SR [[38], table 5, column 6, footnote d).
By this relapse definition, the continuing-care patients who dropped out early and never called in, or the many patients who called in once or more without scoring ≥11 and then dropped out, could never meet the relapse criterion. Because the likelihood of relapse increases with time in follow-up, this definition is biased toward underestimating relapse rates. It is common for patients to lose hope when depressive symptoms return thereby increasing their likelihood of discontinuing a treatment that is no longer effective for them. This is particularly true for the 75% of STAR*D patients diagnosed with ‘recurrent depression’ [[38], table 2]. Given that most STAR*D patients had ‘reoccurring’ depression and ‘loss of hope’ is one of the most common symptoms of depression, it is reasonable to expect that many continuing-care dropouts relapsed.
The summary report of STAR*D identifies 1,854 remitted patients in steps 1–4 [[38], table 3, row 8] yet only 1,518 consented to continuing care [[38], table 5, column 2] while the other 336 dropped out. Many of these patients achieved their remission based on the paper-and-pencil QIDS-SR in their last clinic visit but then discontinued treatment without taking the HRSD despite the USD 25.00 payment for taking said measure (e.g. the step-1 article states, ‘Of the 690 patients with missing exit HRSD scores, 152 (22.1%) achieved QIDS-SR remission at the last treatment visit’ [[32], p. 34]. An additional 344 patients consented to continuing care but then dropped out during the 1st month without ever calling into the IVR system [[38], table 5, column 5]. This indicates that 670 of 1,854 remitted patients (36.7%) of STAR*D dropped out within 1 month of their QIDS-SR remission. Due to its open-label prescribing though, the ultimate outcome for these patients (and all dropouts during any phase) is unclear since patients could continue their treatment without staying in STAR*D by paying for their medications and physician services that heretofore had been free. Given that such a decision required assuming this new cost which could be substantial, particularly for the one third who lacked insurance coverage [[38], table 1], it is unlikely that many STAR*D dropouts continued their treatment on antidepressant medication(s).
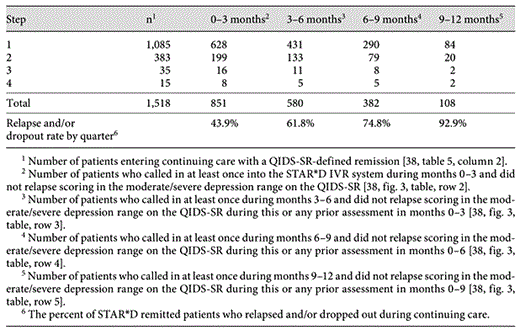
The weighted mean relapse rate of STAR*D for remitted patients that called at least once into its IVR system was 37.4% (range: from 33.5% for step-1 to 50% for step-4 patients) and 64.4% for improved patients who entered continuing care not in remission (range: from 58.6% for step-1 to 83.3% for step-4 patients) [[38], table 5, column 6]. Table 2 presents the survival analysis for the 1,518 patients who entered continuing care in remission. The numbers in table 2 represent the remitted patients who ‘survived’, i.e. did not relapse or dropout. Relapses of course represent unsuccessfully treated patients. Dropouts represent unsuccessfully treated patients as well, viewed from the intent-to-treat perspective. STAR*D authors highlighted their findings that ‘relapse rates were higher for those who entered follow-up after more treatment steps (p< 0.0001)’ [[38], p. 1911] and ‘remission at entry into follow-up was associated with a better prognosis than was simple improvement without remission’ [[38], p. 1912]. While both statements are statistically accurate, they do not address the fact that STAR*D patients’ had extraordinarily high relapse and/or dropout rates during continuing care regardless of the treatment step their remission occurred nor their extent of acute-care improvement prior to entering continuing care.
These findings call into question continuation phase guideline of APA that ‘following remission, patients who have been treated with antidepressant medications in the acute phase should be maintained on these agents to prevent relapse’ despite this recommendation having received the highest ‘clinical confidence’ rating of the expert panel [[40], p. 15].
Discussion
Given its 35 million dollar cost and thoughtful design, STAR*D provides a rare opportunity to evaluate the effectiveness of antidepressants with real-world patients and therefore warrants analysis from multiple perspectives independent of those of its authors. While similar to the analysis of Fava et al. [50] documenting decreasing step-by-step remission rates with increasing rates of relapse and drug intolerance, our analysis found that the results of STAR*D appear even worse than previously realized. Even with the extraordinary care of STAR*D, only about one fourth of patients achieved remission in step 1. The study dropout rate was slightly larger than the success rate. The success rate of step 2 was slightly less than that of step 1 and the dropout rate was larger. The success rates in step 3 (17.8%) and step 4 (10.1%) were even more modest with still larger dropout rates (44.8 and 60.1%, respectively).
Of the 4,041 patients started on citalopram, 370 (9.2%) dropped out within 2 weeks. After up to four trials, each provided with vigorous medication dosing ‘to ensure that the likelihood of achieving remission was maximized and that those who did not reach remission were truly resistant to the medication’ [[32], p. 30], only 1,854 patients (45.9%) obtained remission using the lenient QIDS-SR criteria. In each step, more patients dropped out than were remitted and this dropout rate steadily increased throughout the study. Of the 1,854 remitted patients, 670 (36.7%) dropped out within 1 month of their remission and only 108 (5.8%) survived continuing care and took the final assessment without relapsing and/or dropping out. Even for these 108 patients, it is unclear how many were one of the 607 whose baseline HRSD <14signified only mild symptoms when first started on citalopram in step 1 and therefore had to score worse during continuing care than when they first entered the study to be counted as relapsed.
In STAR*D, failed trials had negative consequences for patients beyond not obtaining remission. Such failures decreased patients’ likelihood for obtaining remission in subsequent trials while increasing their likelihood for drug intolerance, relapse, and/or dropping out. These negative effects lend support to the observation of Fava et al. [[50], p. 262] that successive pharmacological manipulations ‘may propel depressive illness into a refractory phase’ by fostering oppositional tolerance in which the antidepressant sensitizes some patients to depression. Chouinard and Chouinard [[51], p. 75] document similar risks with atypical antipsychotics and estimate that 50% of treatment-resistant schizophrenia cases are related to supersensitivity psychosis. The following issue remains to be determined: the extent that diminishing outcomes of STAR*D are due to a subset developing such oppositional tolerance, patients’ natural diminished expectations of improvement following each failure (i.e. a step-by-step diminishing placebo effect), other unknown factors, and/or some combination thereof.
Most importantly to clinicians, STAR*D results show that antidepressants were only minimally effective with real-world patients when provided consistent with ‘the theoretical principles and clinical beliefs that currently guide the management of treatment-resistant depression’ [[43], p. 136]. This admittedly harsh assessment is most evident when using study completion rates as the best ‘hard measure of treatment effectiveness and acceptability’ [[14], p. 296].
Turner et al. [9] demonstrates how publication bias inflates the perceived efficacy of antidepressants thereby promoting the widespread acceptance of this treatment. The separate analyses of Turner et al. [9] and Kirsch et al. [10] suggest that antidepressants are only marginally efficacious compared to inert placebos, though the findings in these trials may be due to the under-representation of less symptomatic patients with greater comorbidity (particularly anxiety disorders that have been found to lower antidepressant response rates [52]) and long-standing depressive illness who better characterize the majority seeking care. The analysis of Barbui et al. [[15], cited in [16], p. 19] demonstrates how the apparent clinical superiority of paroxetine over placebo disappeared after statistically controlling for differences in drug/placebo side effects suggesting that side effects contribute to unblinding in RCTs and thereby enhance patients’ expectation of improvement since they often guess correctly that they are getting the ‘real’ drug and therefore anticipate improvement. Similar analyses are needed of antipsychotic/placebo antidepressant augmentation trials for ‘treatment-resistant depression’ due to significant side effect profiles of antipsychotics and the role these might play in unblinding. Such analyses are crucial to evaluate properly Nelson and Papakostas’ [53] recent meta-analysis finding that atypical antipsychotics were superior to placebo as augmentation agents since this analysis did not control for said differences in drug/placebo side effects and the fact that this apparent ‘superiority’ was exceptionally modest, with 9 being the number needed to treat to have one additional remission in trials lasting only 6–8 weeks.
What more can we learn from STAR*D and the reviewed articles? First, even with exemplary pharmaceutical efforts it is difficult to achieve sustained recovery for patients reflecting the range of illness severity of STAR*D. Second, the results from efficacy trials (whether for medication, an evidence-based psychotherapy, or any other treatment) are limited in their ability to estimate a treatment benefit to the extent that the ‘the ‘‘apples’’ assessed in such studies do not correspond to the ‘‘oranges’’ of clinical practice’ [26]. The analyses of Zimmerman et al. [30] and Wisniewski et al. [31] further highlight this fundamental disconnect between RCT patients and those most often presenting in real-world clinical practice. Third, the fact that the effectiveness rate in step-2 CTs was no better than antidepressants underscores that MDD – with its common co-morbidities and recurrent nature – is a serious, complex, and difficult-to-treat disorder whose treatment often results in fewer positive outcomes than would be expected from efficacy trials of its two most extensively researched interventions.
Fourth, it is worth considering whether or not the measurement-based system of STAR*D with its focus on measuring side effects and symptoms in every visit until ‘remission’ was achieved hindered or helped patient care. STAR*D authors clearly believed that it helped and even equated ‘high quality care’ with their system stating: ‘Finally, high quality of care was delivered (measurement-based care) with additional support from the clinical research coordinator. Consequently, the outcomes in this report may exceed those that are presently obtained in daily practice wherein neither symptoms nor side-effects are consistently measured and wherein practitioners vary greatly in the timing and level of dosing’ [[38], p. 1914]. STAR*D encouraged all patients who did not achieve remission based on a number to enter the next trial despite the failure of QIDS/HRSD to differentially weight core depressive symptoms (e.g. mood, guilt, suicidal ideation, or anhedonia) and accessory ones (e.g. appetite, insomnia, or agitation) [54,55] and patients’ self-assessments of the relative importance of each. In the absence of a meaningful therapeutic alliance between the patient and doctor, relying instead on patients’ recitation of side effects and symptomatic change to guide treatment, STAR*D may have failed to capitalize on a crucial ingredient necessary for patient improvement. In contrast, psychosomatic methods would likely have improved patient retention and outcomes given its use of more clinically-rich clinimetric assessment procedures, collaborative decision-making, and its focus on enhancing patients’ self-efficacy by teaching them the self-management skills that are likely essential to sustain recovery from MDD [56]. Such psychosomatic methods are at their core ‘psychotherapeutic’ and would likely enhance outcomes from any intervention, be it an antidepressant, a placebo, or some other strategy; particularly when applied to treating depressed – often initially helpless and hopeless – patients and their commonly occurring comorbid conditions.
Fifth, the continuation and maintenance phase guidelines of APA which essentially encourage open-ended use of antidepressants at ‘the same full antidepressant medication doses’ as used in acute treatment appear misguided [[40], p. 15]. While the guidelines of APA are consistent with the meta-analyses performed by Geddes et al. [57] and Papakostas et al. [58] reporting large effect sizes for antidepressants in preventing relapse, these analyses do not control for publication bias [59,60] nor selective outcome reporting [22,23], both of which may significantly inflate the findings from meta-analyses that fail to control for these common forms of researcher bias. For now, prospective naturalistic studies are likely a better guide to estimate real-world outcomes. In STAR*D, even for the 1,085 remitted patients in step 1 who consented to continuing-care and therefore had the highest likelihood of sustained recovery, only 84 (7.7%) did not relapse and/or dropout by the 12th month of continuing care. STAR*D results are similar to findings of Bockting et al. [61] that only 42% used antidepressants continuously during maintenance phase treatment, of whom 60.4% relapsed, whereas patients who stopped using antidepressants experienced less relapse, with only 8% of those who received preventive CT relapsing. These naturalistic continuation phase studies support Fava’s [62,63 ]hypothesis that long-term antidepressant use may worsen the course of depression. Besides these studies, failure to find any apparent benefit from continued antidepressant treatment, the recent finding that long-term use of SSRIs at moderate/high daily doses doubles the risk of diabetes [64], and the uncertain risk of oppositional tolerance, provides additional reasons for reexamining this all too common practice.
Finally, in light of the meager results of STAR*D, it is worth reconsidering the term ‘treatment-resistant depression’ when referring to patients who do not respond favorably to drug treatment. Should we not direct our attention to what is wrong with our treatment rather than classifying some patients as having an exotic form of depression because they fail to respond? Our understanding is hampered by using language that wrongly implies that there is an exceptional subgroup of patients who are refractory to an otherwise effective treatment. The inescapable conclusion from STAR*D results is that we need to explore more seriously other forms of treatment (and combinations thereof) that may be more effective. This effort will require developing new service delivery models to ensure that as such treatments are identified, they are widely implemented [65,66].
Despite the pervasive belief regarding the effectiveness of antidepressants and cognitive therapy (CT) among physicians and society at large, STAR*D shows that antidepressants and CT fail to result in sustained positive effects for the majority of people who receive them. STAR*D authors noted at the outset of the study that the ‘results should have substantial public health and scientific significance’. As healthcare professionals and in line with what STAR*D authors themselves recommend, we should take notice of what this largest antidepressant effectiveness trial ever conducted is telling us and reassess the role of antidepressant medications and CT in the evidence-based treatment for depression.
Acknowledgments
The authors would like to thank both peer reviewers whose insightful comments and suggestions were invaluable in improving the paper.
Conflicts of Interest
H. Edmund Pigott, PhD, and Gregory S. Alter, PhD, are founders of NeuroAdvantage, LLC, a for-profit neurotherapy company. During the past 3 years, Dr. Pigott has consulted for CNS Response, Midwest Center for Stress and Anxiety, and SmartBrain Technologies.
