

Abstract
Mammalian oocytes execute a unique meiotic programme involving 2 arrest stages and an unusually protracted preamble to chromosome segregation during the first meiotic division (meiosis I). How mammalian oocytes successfully navigate their exceptional meiotic journey has long been a question of immense interest. Understanding the minutiae of female mammalian meiosis I is not merely of academic interest as 80–90% of human aneuploidy is the consequence of errors arising at this particular stage of oocyte maturation, a stage with a peculiar vulnerability to aging. Recent evidence indicates that oocytes employ many of the same cast of proteins during meiosis I as somatic cells do during mitosis, often to execute similar tasks, but intriguingly, occasionally delegate them to unexpected and unprecedented roles. This is epitomised by the master cell-cycle regulon, the anaphase-promoting complex or cyclosome (APC/C), acting in concert with a critical APC/C-targeted surveillance mechanism, the spindle assembly checkpoint (SAC). Together, the APC/C and the SAC are among the most influential entities overseeing the fidelity of cell-cycle progression and the precision of chromosome segregation. Here I review the current status of pivotal elements underpinning homologue disjunction in mammalian oocytes including spindle assembly, critical biochemical anaphase-initiating events, APC/C activity and SAC signalling along with contemporary findings relevant to progressive oocyte SAC dysfunction as a model for age-related human aneuploidy.
Introduction
Meiosis is the unique cell division in which chromosome numbers are halved in preparation for sexual reproduction. This contrasts with mitosis, which produces daughter-cells containing the identical chromosomal constitution as the parent-cell. In order to halve the normal chromosome complement, meiosis is comprised of 2 consecutive nuclear divisions following a single round of DNA replication. Meiosis also facilitates genetic diversity by incorporating reciprocal recombination during which genetic material is exchanged between homologous chromosome pairs. Recombination serves the additional function of linking homologues within a structure referred to as a bivalent. During the first meiotic division (meiosis I), bivalents segregate into their component homologues (homologue disjunction) whereas at the second meiotic division (meiosis II), individual homologues divide into 2 sister chromatids reminiscent of mitosis.
In humans, an inextricable link exists between advancing maternal age and declining reproductive success. Thus, older women experience soaring rates of miscarriage and birth defects along with paltry success rates when undergoing assisted reproductive treatments such as in vitro fertilisation (IVF). Aneuploidy is a major contributor to these adverse reproductive outcomes, most often because of errors arising during meiosis I in oocytes, termed non-disjunction errors, the susceptibility to which rises exponentially as women age [Homer, 2007; Vogt et al., 2008; Hassold and Hunt, 2009].
A better understanding of female mammalian meiosis I is therefore of paramount importance for gaining insight into the origins of human reproductive catastrophe. For a number of reasons, access to human oocytes for research is extremely limited, so the vast majority of recent insight has come from work on the well-characterised murine model. Therefore, although this review will refer to data obtained from human oocytes where possible, much of the ensuing discussion draws on work undertaken in mouse oocytes.
Overview of Female Mammalian Meiosis
The timeline of meiosis in female mammals is very protracted and punctuated by 2 arrest phases. In females, recombination occurs during fetal life after which oocytes undergo their first arrest at the dictyate stage of prophase I. Such oocytes are identifiable by the presence of an intact nucleus referred to as the germinal vesicle (GV). GV-arrest can last decades in humans and is only broken postnatally by hormonal cues arising from puberty onwards. Resumption of meiosis I is marked by GV breakdown (GVBD) following which oocytes segregate recombined homologous chromosomes before undergoing first polar body extrusion (PBE), the morphological marker of exit from meiosis I (fig. 1). Oocytes are then arrested for a second time at metaphase of meiosis II (metaphase II) under the influence of an ooplasmic activity referred to as cytostatic factor. The focus of this chapter will be meiosis I which, for the purposes of this discussion, will be used to refer to the stage from GVBD to PBE (fig. 1).
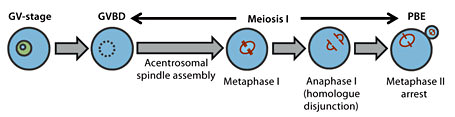
Schematic of female mammalian meiosis I. Mammalian oocytes arrested at the dictyate stage of prophase I are identifiable by the presence of a germinal vesicle (GV). Resumption of meiosis I is marked by GV breakdown (GVBD) following which bivalents are brought to alignment at the spindle equator by metaphase I. Anaphase I then ensues when chromosomes segregate between the secondary oocyte and the polar body. Following first polar body extrusion (PBE), oocytes progress without a hiatus into meiosis II where they are are arrested for a second time at metaphase II.
Schematic of female mammalian meiosis I. Mammalian oocytes arrested at the dictyate stage of prophase I are identifiable by the presence of a germinal vesicle (GV). Resumption of meiosis I is marked by GV breakdown (GVBD) following which bivalents are brought to alignment at the spindle equator by metaphase I. Anaphase I then ensues when chromosomes segregate between the secondary oocyte and the polar body. Following first polar body extrusion (PBE), oocytes progress without a hiatus into meiosis II where they are are arrested for a second time at metaphase II.
In mammalian oocytes, the interregnum between GVBD and PBE is extraordinarily drawn out, lasting around 10 h in mouse oocytes and a staggering 36 h or so in human oocytes. In stark contrast, in HeLa cells, one of the most frequently used experimental somatic cell types, the interval from nuclear envelope breakdown (NEBD) to anaphase is about 30 min in the majority of instances [Meraldi et al., 2004]. In oocytes, a considerable proportion of their hours-long meiosis I is devoted to sculpting a bipolar spindle.
Acentrosomal Spindle Assembly
The spindle is the cellular infrastructure comprised of microtubules that mechanically segregates chromosomes. Unlike mitosis in which spindle poles and hence spindle bipolarity are predetermined by 2 microtubule-nucleating centrosomes, during female mammalian meiosis I, spindles are assembled without centrosomes. In mouse oocytes, spindle morphology undergoes quite dramatic changes from a spherical microtubule ‘ball’ shortly after GVBD (fig. 2A) to the mature bipolar spindle (fig. 2B) some hours later. This microtubule metamorphosis is critical for ensuring that chromosomes ultimately become securely anchored to the spindle in the correct configuration prior to the onset of anaphase I (fig. 2C) when one member of each pair of homologues is delivered either to the secondary oocyte or to the vestigial first polar body.

Chromosome and spindle dynamics during acentrosomal spindle assembly in mouse oocytes. Oocytes of the MF1 strain of mouse were immunostained for kinetochores (CREST), chromosomes (DNA) and spindle microtubules at 2 h post-GVBD (A), 8 h post-GVBD (B) and 9 h post-GVBD (C). Note the ball-like microtubule configuration in early prometaphase I (A) which transforms into a barrel-shaped bipolar spindle by metaphase I (B).
Chromosome and spindle dynamics during acentrosomal spindle assembly in mouse oocytes. Oocytes of the MF1 strain of mouse were immunostained for kinetochores (CREST), chromosomes (DNA) and spindle microtubules at 2 h post-GVBD (A), 8 h post-GVBD (B) and 9 h post-GVBD (C). Note the ball-like microtubule configuration in early prometaphase I (A) which transforms into a barrel-shaped bipolar spindle by metaphase I (B).
Understanding the mechanisms by which the oocyte moulds a bipolar spindle is therefore important for understanding the overall execution of homologue segregation. It was recently shown that the spindle in mouse oocytes is derived from over 80 microtubule organising centres (MTOCs) which arise de novo from a cytoplasmic microtubule repository [Schuh and Ellenberg, 2007]. MTOC-driven microtubule proliferation is dependent on TPX2 (Targeting Protein for the Xenopus kinesin xklp2) as oocytes depleted of the majority of TPX2 assemble very rudimentary spindles having markedly reduced microtubule density [Brunet et al., 2008]. As meiosis I advances, MTOCs are progressively focused into 2 poles under the action of the tetrameric plus-end-directed motor, kinesin-5, inhibition of which robustly blocks spindle assembly at the microtubule ball stage [Schuh and Ellenberg, 2007]. Subsequent maintenance of spindle pole integrity requires the action of TACC3 (Transforming Acidic Coiled Coil protein 3) likely in its phosphorylated form (P-TACC3), as P-TACC3 and not TACC3 increases progressively during meiosis I, P-TACC3 becomes enriched at spindle poles and TACC3-depleted oocytes show spindle defects [Brunet et al., 2008]. The production of P-TACC3 is itself dependent upon the N-terminal region of TPX2 [Brunet et al., 2008], TPX2 probably acting via Aurora A kinase as the N-terminal domain of TPX2 has been shown to activate Aurora A kinase, which in turn is known to phosphorylate and activate TACC3.
TPX2 is a recognised target of the Ran pathway involving the small GTPase Ran in its GTP-bound form (Ran-GTP). In many systems, Ran-GTP is concentrated in the region of chromosomes where it releases TPX2 and other spindle assembly factors from inhibitory sequestration by importins. By augmenting such factors in the region of chromosomes, it is believed that the Ran-GTP gradient is integral to the overall efficiency of spindle assembly. Recently, Ran-GTP was shown to concentrate in the region of chromosomes in mouse oocytes [Dumont et al., 2007] and to promote the initial upsurge in microtubule mass occurring during the first couple of hours following GVBD [Schuh and Ellenberg, 2007]. Interestingly however, Ran-dependence in early prometaphase I appears to be largely superseded by Ran-independent factors later in meiosis I as oocytes in which Ran-GTP generation is inhibited using a mutant form of Ran (RanT24N) are still capable of assembling bipolar spindles, albeit with delayed kinetics [Dumont et al., 2007; Schuh and Ellenberg, 2007]. Moreover, spindles assembled in the presence of RanT24N are capable of accurately segregating homologous chromosomes [Dumont et al., 2007]. Ran-independent factors for spindle assembly appear to be nuclear-derived as spindle assembly is completely disrupted in enucleated oocytes in which Ran-GTP is inhibited [Schuh and Ellenberg, 2007]. It has been suggested that the very extended duration of meiosis I might render Ran-GTP superfluous as there is ample time for alternative pathways, albeit less efficient, to complete spindle assembly prior to PBE [Dumont et al., 2007]. In line with this, Ran-GTP is indispensable when spindle assembly occurs within much shorter time frames such as during meiosis II when spindles assemble within 1 h of PBE [Dumont et al., 2007].
As spindle assembly proceeds, chromosomes ultimately become properly aligned at the spindle equator (fig. 2B) by attaining stable attachments to both spindle poles. Such bipolar attachment depends upon the formation of microtubule bundles anchored in an end-on manner to kinetochores called K-fibres. Kinetochores are multiprotein complexes assembled on centromeric DNA that serve as the interface between chromosomes and microtubules (see fig. 2). Electron microscopic examination of oocytes could only identify K-fibres in oocytes that were fixed many hours following GVBD suggesting that K-fibre formation is a relatively late event during meiosis I [Brunet et al., 1999]. Notably however, although electron microscopy has provided some insight into the timing of K-fibre establishment, more dynamic facets of kinetochore-microtubule interactions and the key proteins required for K-fibre formation within the context of the mammalian oocyte remain largely unknown.
In order to avert aneuploidy, it is vital that cells are fastidious in ensuring that the biochemical events that execute anaphase are licensed only after kinetochore-microtubule interactions have been completed. As discussed later, the responsibility for coordinating these 2 processes lies with a fail-safe mechanism known as the spindle assembly checkpoint (SAC).
Conducting Anaphase
The Anaphase-Promoting Complex
The biochemical events underpinning anaphase-onset are mediated by a master cell-cycle regulon aptly known as the anaphase-promoting complex or cyclosome (APC/C), a multimeric ubiquitin ligase [Pesin and Orr-Weaver, 2008]. The APC/C in conjunction with 1 of 2 co-activators, Cdc20 or Cdh1, polyubiquitinates substrates thereby earmarking them for destruction by the 26S proteasome.
APC/C activated by either Cdc20 (APC/CCdc20) or Cdh1 (APC/CCdh1) targets substrates via recognition motifs known as destruction boxes, the 2 best known being the D-box and the KEN-box. The D-box is recognisable by either APC/C species whereas the KEN box is exclusively targeted by APC/CCdh1. In this way, the APC/C orchestrates critical cell-cycle transitions by directing the timely destruction of key proteins.
Bringing About Anaphase in Somatic Cells
During mitosis, APC/CCdc20 triggers the metaphase-to-anaphase transition. APC/CCdc20 mediates the destruction of 2 pivotal proteins, securin and cyclin B, the latter resulting in inactivation of cyclin-dependent kinase 1 (CDK1, also known as maturation-promoting factor or MPF in oocytes). Securin proteolysis and CDK1 inactivation together result in the liberation of the protease, separase. Active separase in turn cleaves SCC1/RAD21/MCD1, the α-kleisin subunit of a molecular ‘glue’ tethering duplicated chromosomes together known as the cohesin complex. This culminates in loss of cohesion between replicated chromosomes, so permitting them to be drawn to opposing poles by spindle microtubules at anaphase.
APC/CCdc20 Mediates Anaphase I-Onset in Mammalian Oocytes
Until relatively recently, very little had been known about the mechanisms responsible for anaphase I-onset or whether or not the APC/C was involved in meiosis I progression in vertebrate oocytes. In mouse oocytes, it had long been known that MPF inactivation accompanies exit from meiosis I and that this is the consequence of cyclin B destruction [Hampl and Eppig, 1995], suggesting a role for the APC/C. However, this reasoning was brought into question by the surprising finding that the APC/C appeared dispensable for the meiosis I-to-meiosis II transition in another vertebrate model system, Xenopus oocytes [Peter et al., 2001; Taieb et al., 2001].
Subsequent work specifically addressed the requirement of APC/C activity during meiosis I in mammalian oocytes. A powerful experimental tool has been the use of fluorescently-tagged constructs of securin and cyclin B. These fluorescent versions of securin and cyclin B are considered faithful reporters for their endogenous counterparts [Clute and Pines, 1999] enabling securin and cyclin B turnover to be monitored in live oocytes during progression though meiosis I. Using these fluorescent chimerae, it was shown that securin and cyclin B undergo net synthesis following GVBD and decline precipitously on exit from meiosis I [Herbert et al., 2003]. Western blotting subsequently corroborated these data and showed that endogenous securin did indeed undergo destruction on exit from meiosis I akin to cyclin B [Hampl and Eppig, 1995; Reis et al., 2007; Homer et al., 2009]. The decline of securin and cyclin B in mouse oocytes indirectly alluded to APC/C participation at the meiosis I-to-meiosis II transition.
Direct evidence for APC/C involvement in meiosis I was very recently reported using an elegant approach in which Cre recombinase was used to delete a floxed allele of the APC2 gene, encoding one of the subunits of the APC/C core [McGuinness et al., 2009]. Notably, because Cre recombinase was under the control of the Zona pellucida 3 (Zp3) promoter, it was expressed exclusively during the oocyte’s growth stages. Thus, any effects observed in apc2Δ/Δ oocytes could be attributed to meiosis-specific defects brought about through loss of APC/C activity and would not be confounded by possible hang-over effects arising earlier during pre-meiotic mitotic divisions. Using fluorescently-labelled securin, it was found that although apc2Δ/Δ oocytes synthesised securin post-GVBD, they were incapable of mediating securin destruction, segregating homologues or undergoing PBE, altogether confirming an indispensable role for the APC/C in modulating anaphase I [McGuinness et al., 2009].
Other work addressed which APC/C species, APC/CCdc20 or APC/CCdh1, is responsible for the metaphase I-to-anaphase I transition in mouse oocytes. A destruction box-mutated securin construct resistant to APC/CCdc20-directed destruction but susceptible to APC/CCdh1 was found to be stable in oocytes and its expression was accompanied by failure of homologue disjunction and inhibition of PBE [Herbert et al., 2003]. Added to this, in oocytes in which Cdc20 was depleted using morpholino oligonucleotides, securin and cyclin B destruction and PBE were inhibited whereas inhibition of protein destruction and PBE were not features of Cdh1-depleted oocytes [Reis et al., 2007]. Collectively, these data indicated that in mammalian oocytes, APC/CCdc20 and not APC/CCdh1 is required for the metaphase I-to-anaphase I transition and PBE through the destruction of securin and cyclin B.
Resolving Cohesion: Separase and REC8
From the foregoing, it is clear that APC/CCdc20-dependent securin and cyclin B destruction constitutes the driving force for separating chromosomes during meiosis I in mouse oocytes. Other critically important pieces of the puzzle pertain to the roles of separase and cohesin. Notably, these roles must accommodate the unique step-wise release of cohesion, first elucidated in the yeast model, necessary for conducting 2 sequential nuclear divisions during meiosis. Thus, whereas mitotic anaphase requires that all cohesins be dissolved, cohesion resolution during meiosis I must be discriminatory so that only arm cohesion is lost at anaphase I, leaving centromeric cohesion to be removed during anaphase II (fig. 3). The proper regulation of centromeric cohesion is critically dependent upon a recently described centromeric guardian, shugoshin, the subject of accompanying chapters in this issue.

Schematic of homologue disjunction in mouse oocytes. Shown is a schematic representation of a murine bivalent compromised of 2 homologous chromosomes (blue and green) which have undergone recombination. Each homologous chromosome is comprised of 2 sister chromatids. Homologue disjunction during meiosis I is induced by selective loss of arm cohesion (brown) following which sister chromatids remain tethered by centromeric cohesion (yellow; A). This residual centromeric cohesion is then resolved at anaphase II when sister chromatids separate (B). Note also that during meiosis I, sister kinetochores (grey ovals) of each homologue attach to microtubules (black lines) emanating from the same pole (syntelic attachment) whereas in meiosis II, sister kinetochores now become attached to opposite poles (amphitelic attachment).
Schematic of homologue disjunction in mouse oocytes. Shown is a schematic representation of a murine bivalent compromised of 2 homologous chromosomes (blue and green) which have undergone recombination. Each homologous chromosome is comprised of 2 sister chromatids. Homologue disjunction during meiosis I is induced by selective loss of arm cohesion (brown) following which sister chromatids remain tethered by centromeric cohesion (yellow; A). This residual centromeric cohesion is then resolved at anaphase II when sister chromatids separate (B). Note also that during meiosis I, sister kinetochores (grey ovals) of each homologue attach to microtubules (black lines) emanating from the same pole (syntelic attachment) whereas in meiosis II, sister kinetochores now become attached to opposite poles (amphitelic attachment).
Initial investigations into the role of separase in mouse oocytes sought to inhibit separase activity. One approach involved a separase inhibitor purported to covalently bind the enzyme’s active site [Terret et al., 2003]. Microinjection of the separase inhibitor induced high levels of spindle and chromosome aberrations including chromosomes lagging between spindle poles after anaphase I suggesting that separase is required for efficient homologue disjunction [Terret et al., 2003]. Another approach sought to prevent separase activation by maintaining high levels of securin, either by over-expressing the wild-type protein [Terret et al., 2003] or by expressing a mutant form of securin resistant to APC/C-mediated destruction [Herbert et al., 2003]. Both interventions were found to prevent homologue disjunction, thereby indirectly implicating separase in meiosis I chromosome segregation.
Subsequently, separase-deficient oocytes (SeparaseΔ oocytes) were generated using Zp3 promoter-driven Cre-recombinase to delete exons encoding separase’s protease domain in growing oocytes [Kudo et al., 2006] as described above for deleting APC2. As judged by time-lapse imaging of fluorescently labelled chromosomes as well as chromosome spreads, homologue disjunction was found to be robustly inhibited in SeparaseΔ oocytes providing incontrovertible evidence that separase is indispensable for chiasma resolution during female meiosis I.
During meiosis, a cohesin variant containing the meiosis-specific α-kleisin, REC8, confers cohesion within bivalents. In yeast, separase-mediated cleavage of REC8 removes cohesin from chromosome arms and is required for homologue disjunction [Buonomo et al., 2000]. The requirement for separase during meiosis I in mouse oocytes suggested that separase-mediated REC8 cleavage might be a conserved process for segregating homologues. In keeping with this, immunostaining showed that REC8 along chromosome arms is lost after anaphase I in wild-type mouse oocytes but not in SeparaseΔ oocytes [Kudo et al., 2006; Lee et al., 2006]. Thus, in mouse oocytes, loss of REC8 from chromosome arms accompanies homologue disjunction and is dependent upon separase.
A recent paper examined whether the observed separase-dependent loss of REC8 from chromosome arms in mouse oocytes was the consequence of REC8 cleavage [Kudo et al., 2009]. Three separase-targeted cleavage sites were identified in mouse REC8 and mutated to produce a construct (REC8-N) that was resistant to separase-mediated cleavage in vitro. A transgenic mouse was engineered to express REC8-N by microinjecting pronuclear stage embryos with a bacterial artificial chromosome harbouring the sequence encoding REC8-N. On the basis that a non-cleavable REC8 protein blocks homologue disjunction in yeast even when co-expressed with the endogenous wild-type REC8 [Buonomo et al., 2000], it might be predicted that homologue disjunction would be similarly blocked in transgenic REC8-N mouse oocytes. Somewhat surprisingly, however, the phenotype observed was relatively subtle as homologue disjunction and PBE rates were comparable between transgenic and wild-type oocytes with the result that fertility was not greatly compromised in transgenic REC8-N females. Notably however, when compared with wild-type oocytes, there was a delay of about 2 h between the time securin was almost completely destroyed (a surrogate marker for separase activation) and the completion of homologue segregation in transgenic oocytes. This was interpreted as signifying that cleavage of REC8-N was less efficient than that of wild-type REC8. Although possibly slowed down, REC8-N cleavage was not abolished, as immunostaining of chromosome spreads revealed that REC8-N was present during meiosis I and largely lost from chromosome arms by meiosis II. The authors proposed that cryptic cleavage sites in REC8-N may have rendered it vulnerable to separase-mediated cleavage, albeit with lowered efficiency. Thus, it appears that REC8 cleavage is required for efficient cohesin removal during meiosis I. However, it is unclear whether separase-mediated cleavage is absolutely necessary for ultimately resolving chiasmata in mammalian oocytes.
A Reversal of the Mitotic Paradigm: APC/CCdh1 before APC/CCdc20 in Oocytes
Prior to mitotic anaphase, APC/C cannot be activated by Cdh1, as CDK1-induced inhibitory phosphorylation precludes Cdh1’s interaction with the APC/C holoenzyme [Pesin and Orr-Weaver, 2008]. As CDK1 activity falls following anaphase-onset, however, APCCdh1 activity rises whereupon it continues the destruction of securin and cyclin B previously initiated by APC/CCdc20. Thus, in mitosis, APC/CCdc20 is the APC/C species which is active first as the anaphase-initiator prior to APC/CCdh1 in late mitosis.
It seemed as though APC/C activity in mammalian oocytes might replicate the mitotic blueprint until it was discovered that APC/CCdh1 is active in oocytes during prophase I where it sustains the arrest-state by restraining cyclin B accumulation and hence MPF activation needed for GVBD [Reis et al., 2006]. It was subsequently demonstrated that APC/CCdh1 activity perdures after GVBD into prometaphase I where it degrades Cdc20 [Reis et al., 2007]. This APC/CCdh1-directed Cdc20 proteolysis is essential for preventing premature APC/CCdc20 activation, which would otherwise induce early exit from meiosis I along with marked aneuploidy [Reis et al., 2007]. As discussed in greater detail below, the APC/CCdh1 saga in oocytes continues to evolve as we recently demonstrated that along with Cdc20, APC/CCdh1 also degrades securin during early prometaphase I [Homer et al., 2009].
Intriguingly therefore, APC/CCdh1 is active prior to APC/CCdc20 during meiosis I in mouse oocytes (fig. 4) contrasting sharply with the mitotic model in which the exact reverse occurs with APC/CCdh1 following on the heels of APC/CCdc20. Thus, unlike mitotic prometaphase in which there is primarily one active APC/C species (activated by Cdc20), during prometaphase I, the oocyte is tasked with modulating 2 APC/C species ahead of anaphase I (fig. 4). An important issue regarding the integrity of homologue disjunction therefore, is how these 2 potent APC/C species are marshalled.

Schematic of APC/C activities during meiosis I in mouse oocytes. APC/CCdh1 is active during prophase I arrest and early prometaphase I prior to APC/CCdc20 during late meiosis I. The SAC modulates APC/CCdc20 activity thereby delaying anaphase I-onset so as to allow sufficient time for completion of kinetochore-microtubule attachments. The periods of APC/C and SAC activities are approximations.
Schematic of APC/C activities during meiosis I in mouse oocytes. APC/CCdh1 is active during prophase I arrest and early prometaphase I prior to APC/CCdc20 during late meiosis I. The SAC modulates APC/CCdc20 activity thereby delaying anaphase I-onset so as to allow sufficient time for completion of kinetochore-microtubule attachments. The periods of APC/C and SAC activities are approximations.
The Spindle Assembly Checkpoint: Keeping APC/CCdc20 in Check
Since APC/CCdc20 licenses the irreversible commitment to chromosome separation, its activity must be meticulously suppressed until such time that the risk of chromosome mis-segregation (and hence aneuploidy) is averted. The responsibility for policing the APC/CCdc20 rests squarely with the SAC, the principal components of which are derived from members of the BUB (Budding Uninhibited by Benzimidazole) and MAD (Mitotic Arrest-Deficient) protein families [Musacchio and Salmon, 2007].
At the hub of SAC signalling is the kinetochore; the prevailing consensus is that the kinetochore is the platform from which the SAC orchestrates proceedings. The kinetochore is an optimal vantage point for SAC signalling as this is where attachments between chromosomes and microtubules will form and it is these kinetochore-microtubule attachments that must be of a specific configuration if high-fidelity chromosome segregation is to follow. Under conditions in which kinetochores are unattached or mal-attachments exist therefore, the SAC delays anaphase-onset by inhibiting APC/CCdc20 until such time that correct attachments are established [Musacchio and Salmon, 2007].
Given that APC/CCdc20 drives the metaphase I-to-anaphase I transition in oocytes, the prediction is that an SAC would also be required for properly regulating the timing of homologue disjunction. However, the SAC in mammalian oocytes has a checkered past and has undergone a 180-degree turnaround in a little over 2 decades. The earliest reports regarding SAC function were intuited based on observations in oocytes derived from mutant mice monosomic for the X chromosome (XO oocytes) [LeMaire-Adkins et al., 1997]. XO oocytes were observed to progress through meiosis I without delay, indeed, they appeared to do so at an accelerated schedule, apparently in spite of the presence of a misaligned achiasmate X chromosome in many instances [LeMaire-Adkins et al., 1997]. This was counter to the principles of a robust SAC-monitored system in which anaphase should be delayed by any such alignment aberration. On the basis of this, it seemed that mammalian oocytes might lack this important surveillance mechanism during meiosis I. Such a deficiency provided an appealing mechanism by which human oocytes would be vulnerable to high levels of non-disjunction, an observation already well-established but in search of a mechanistic aetiology.
Subsequent interrogation of the SAC sought to address the fundamental principles of SAC signalling in normosomic strains of mice. One of the cornerstones of SAC investigation is the enduring principle that an SAC-competent cell should either arrest or delay cell-cycle progression in response to disrupted kinetochore-microtubule attachments; experimentally, the commonest means for artificially perturbing kinetochore-microtubule interactions is by treating cells with spindle poisons such as nocodazole and taxol. One of the first systematic investigations of SAC function during female meiosis I examined the response of mouse oocytes to nocodazole treatment [Wassmann et al., 2003]. It was found that low doses of nocodazole delayed progression through meiosis I. Furthermore, a dominant negative MAD2 mutant was found to override this delay implicating the SAC in mediating the observed response.
At this stage, the critical unresolved question pertained to whether or not an SAC was necessary in unperturbed oocytes for safeguarding the integrity of homologue disjunction. This was addressed using a morpholino-based approach to deplete ∼80% of MAD2 in fully-grown mouse oocytes [Homer et al., 2005c]. It should be noted that prior to the use of post-transcriptional gene silencing tools in oocytes, the more conventional approach based on gene knockout animals was uninformative regarding oocyte SAC function as homozygous null SAC mutants are uniformly embryonic lethal. Oocytes depleted of MAD2 using morpholinos suffered a striking increase in aneuploidy, establishing for the first time that mammalian oocytes rely on an SAC for accurate homologue disjunction [Homer et al., 2005c]. Heightened aneuploidy in metaphase II-stage oocytes was accompanied by premature securin and cyclin B destruction during meiosis I and accelerated PBE [Homer et al., 2005c]. Together these data indicated that non-disjunction in MAD2-depleted oocytes was due to precocious anaphase-initiation. Thus, the SAC is required during female mammalian meiosis I for accurate chromosome segregation by delaying anaphase I-onset.
Subsequent studies have investigated additional SAC components such as BUB3 [Li et al., 2009] and BUB1 [McGuinness et al., 2009] amongst others and have reaffirmed the indispensability of the SAC for arresting meiosis I during chemical exposure and/or for averting non-disjunction by preventing premature anaphase I-onset.
APC/CCdh1 and a New Signalling Paradigm
Whereas APC/CCdc20 activity in oocytes and its modulation by the SAC in many ways resemble the mitotic template, APC/CCdh1 activity during prometaphase I in oocytes is uncharted waters. Nevertheless, insight into oocyte-based control of this potent proteolytic machine is gradually evolving.
The Mammalian Oocyte Provides a Permissive Environment for APC/CCdh1 Activity during Early Meiosis I
At NEBD in mitotic cells, phosphorylation-dependent Cdh1 inhibition is already enforced, thereby precluding its association with APC/C [Pesin and Orr-Weaver, 2008]. In contrast, in oocytes, the slow rate of cyclin B accumulation following GVBD means that MPF remains below the threshold required for Cdh1 inhibition until some hours into meiosis I [Reis et al., 2007]. In mitosis, EMI1 (Early Mitotic Inhibitor 1) is a potent inhibitor of APC/CCdc20 during prophase but not in prometaphase due to its destruction following NEBD. Although EMI1 also regulates prophase I in mouse oocytes, here APC/CCdh1 is its main focus [Marangos et al., 2007]. Notably however, EMI1 may not influence APC/CCdh1 activity during prometaphase I in oocytes as it is rapidly degraded following GVBD [Marangos et al., 2007]. Thus, attenuated Cdh1 inhibitory phosphorylation in combination with an absence of EMI1 provide at least 2 reasons why early prometaphase I in oocytes is permissive for APC/CCdh1. Eventually, APC/CCdh1 is squelched during the latter phases of prometaphase I when cyclin B levels and consequently MPF activity surpass the threshold for Cdh1 inhibition along with which Cdh1 is degraded, possibly at the hands of APC/CCdh1 itself [Reis et al., 2007].
A bona fide APC/CCdc20 Inhibitor Doubles as an APC/CCdh1 Promoter
Cdc20 was the first prometaphase I substrate of APC/CCdh1 to be identified in oocytes [Reis et al., 2007]. Our recent data uncovered another important substrate during early prometaphase I, securin [Homer et al., 2009]. This was an incidental discovery whilst investigating the role of a key SAC component, BUBR1 (BUB-Related 1).
In somatic cells, BUBR1’s canonical role is as a core component of the SAC required for efficient APC/CCdc20 inhibition [Musacchio and Salmon, 2007]. In oocytes, however, it turned out that BUBR1 possessed a previously unreported and unexpected regulatory dimension aimed at Cdh1. Specifically, we found that BUBR1 was required for stabilising Cdh1 as endogenous Cdh1 levels were 60–70% lower in oocytes depleted of ∼80% of BUBR1 [Homer et al., 2009]. This brings us back to APC/CCdh1-directed securin destruction as BUBR1-depleted oocytes accumulated 2-fold higher securin levels as a consequence of reduced Cdh1 and consequently, diminished APC/CCdh1 activity. Moreover, this securin over-accumulation overwhelmed later-acting APC/CCdc20 effectively culminating in relative securin stabilisation and hence in an inability to segregate homologues presumably because of the failure to activate separase. Thus, the phenotype of oocytes depleted of the majority of BUBR1 is prometaphase I arrest.
Intriguingly therefore, APC/CCdh1-directed securin proteolysis is essential for meiosis I progression but in the opposite direction from APC/CCdh1-dependent Cdc20 destruction. That is, whereas compromised APC/CCdh1-dependent Cdc20 destruction leads to premature APC/CCdc20 activation and early meiosis I exit [Reis et al., 2007], loss of APC/CCdh1-mediated securin destruction in early meiosis I culminates in a pervasive meiotic arrest [Homer et al., 2009].
It is important to note that these findings in no way exclude a more traditional APC/CCdc20-directed SAC role for BUBR1 in oocytes. In keeping with this, BUBR1 localises to kinetochores and over-expression of 2 mutant forms of BUBR1 lacking an important CDC20-binding domain required for SAC activity accelerates meiosis I by ∼2 h [Tsurumi et al., 2004; Homer et al., 2009]. This was reminiscent of the phenotype produced by depletion of MAD2 [Homer et al., 2005c], pointing to an SAC function for BUBR1. In oocytes therefore, BUBR1 could be a master-regulator, first, by sustaining prometaphase I progression through a positive influence on APC/CCdh1 and second, by negatively regulating APC/CCdc20 activity within the context of the SAC. Notably, both functions are essential for anaphase I regulation albeit via strikingly different mechanisms.
These data introduce a new genre in cell-cycle regulation in which an active mechanism is required for sustaining forward progression. Until now, the focus has predominantly been on mechanisms (epitomised by the SAC) which delay cell-cycle advance thereby furnishing sufficient time for proper spindle assembly and chromosome alignment. Our data suggest that perhaps because of their extended hours-long meiosis I, mammalian oocytes are at risk of stalling due to deregulation of critical cell-cycle proteins and must engage active mechanisms for keeping them in check. Indeed, oocytes arrested in meiosis I (so-called maturation failure) is a recognised condition in IVF programmes in spite of supra-physiological levels of hormonal ovarian stimulation [Beall et al., 2010].
Does SAC Dysfunction Contribute to Age-Related Aneuploidy?
There is now incontrovertible evidence for the existence of an SAC in mammalian oocytes. The next issue of over-arching importance pertains to whether progressive oocyte SAC dysfunction contributes to the exponential rise in human aneuploidy observed with advancing female age.
Modelling Human Aneuploidy in the Mouse
The most direct means for proving or disproving a role for the SAC in age-related human aneuploidy, interrogating human oocytes, is plagued by considerable bioethical and technical impediments. An alternative approach is to use an animal model such as the mouse. However, whereas in the mouse, female reproductive function declines after only 1 to 2 years, tangible declines in the reproductive performance of women do not generally occur before 30 years or more. Furthermore, some may contend that aneuploidy rates and more importantly, age-related increases, in mice do not reflect the human situation. Understandably therefore, skepticism abounds as to whether the mouse is a credible surrogate for addressing the human biological phenomenon.
A very recent and comprehensive study sought to resolve this issue by asking whether natural aging in the mouse might be associated with key hallmarks of human reproductive aging [Pan et al., 2008]. In this work, the mouse strains used (B6D2F1 and B6SJLF1 strains) have a life expectancy of 132 weeks. On the basis of a human female life expectancy of ∼84 years, it was extrapolated that 60–70-week-old mice would correspond with women aged 38–45 years. B6D2F1 and B6SJLF1 mice exhibited a marked decline in ovulation with advancing age and a steep rise in meiosis I chromosome segregation errors from ∼8% at 8 weeks of age to a staggering 50% at 70 weeks, akin to the trends observed in women. Together, these data constitute some of the most robust support for the mouse as a working model for studying human aneuploidy.
Gene expression profiling of human and mouse oocytes provide other important parallels between the 2 systems. Globally, total numbers of transcripts in mouse and human oocytes are very similar (about 10,000 to 11,000) [Hamatani et al., 2004; Grøndahl et al., 2010]. Additionally, in both, a relatively small proportion of the transcriptome (∼4–5%) is differentially expressed with age [Hamatani et al., 2004; Grøndahl et al., 2010]. Notably, in both mouse and human oocytes, transcripts related to cell-cycle pathways are some of the most vulnerable to aging and, amongst the cell-cycle genes, SAC transcripts feature prominently.
Female Reproductive Outcome in Mutant Mice
The disruption to meiosis I chromosome segregation observed when SAC genes are experimentally silenced in oocytes combined with age-related changes in oocyte SAC transcript levels (discussed later) support the notion that SAC dysfunction in oocytes could be a major contributor to age-related increases in female meiosis I non-disjunction. Whilst this correlation provides a platform for an extremely appealing model, it does not on its own constitute formal proof of principle. To explore this further, it will be instructive to examine the evidence pertinent to this hypothesis and whether key predictions embodied within it are borne out by the data.
A central prediction for any working SAC-based model is that animals bearing targeted mutations in SAC genes would replicate key elements of reproductive performance observed for older women; that is, mutant mice should undergo increased rates of meiosis I chromosome mis-segregation at the oocyte-level and this in turn would impair fertility at the organism level. In support of this, oocytes from mutant female mice with only a single functional Mad2 allele (Mad2+/– mice) experience higher rates of aneuploidy (∼22%) when compared with control oocytes (∼5%) [Niault et al., 2007]. Furthermore, female Mad2+/– mice experience reproductive problems as 21% are sterile and amongst those females that are fertile, there is a 42% net reduction in litter size. Interestingly, this effect is gender specific as fertility does not appear to be compromised in Mad2+/– male mice. Mutant mice expressing reduced levels of BUBR1 are also sub-fertile although in this instance, the fertility insult is not gender-specific [Baker et al., 2004].
In the aforementioned mouse mutants, reproductive compromise is the consequence of reduced levels of SAC proteins, neatly supporting the contention that aging could impair SAC function simply by leading to a reduction in the levels of SAC components. However, 2 recent studies illustrate that SAC defects can arise by means other than diminished SAC protein levels.
In one study, mice heterozygous for a truncated version of BUB1 (Bub1+/m mice) were generated using a knockin gene-trap mutation within the BUB1 gene. Although these mice expressed both full-length BUB1 (from the wild-type allele) and a truncated BUB1 variant (from the mutated allele), the latter appeared to compromise the function of the former, and resulted in reduced fertility [Leland et al., 2009]. There was a female gender-specific susceptibility to significantly reduced litter sizes in Bub1+/m mice (as seen with Mad2+/– mice), which was aggravated by advancing age culminating in premature sterility. Notably, the authors traced the cause of age-related female sub-fertility to meiosis I-derived aneuploidy. Moreover, the rates and patterns of aneuploidy in oocytes correlated strongly with that in zygotes, pointing to inherited oocyte-derived aneuploidy as the primary cause of low embryonic viability. Overall, apart from premature sterility, the reproductive behaviour of this SAC mutant mouse very closely mirrors natural human female reproductive aging. This prompted the authors to propose that age-related accumulation of mutations in key SAC genes could contribute to age-related rises in aneuploidy.
The second study involved p73, a member of the p53-related family, which, along with their better known roles as tumour suppressors are also implicated in embryogenesis. In this paper, a mutant mouse (TAp73–/– mice) lacking the TAp73 isoform of p73 (encoded by Trp73) was generated; both male and female TAp73–/– mice were found to be infertile [Tomasini et al., 2008]. Notably, TAp73–/– oocytes exhibited high rates of disordered spindle assembly and following IVF, there was a marked reduction in blastocyst development (28% compared with 75% for wild-type oocytes). Given that older women exhibit a similar spectrum of oocyte and embryonic defects, the authors hypothesised that TAp73 decline could contribute to age-related fertility decline. Indeed, it was further demonstrated that TAp73 levels decline precipitously with natural aging in mouse oocytes [Tomasini et al., 2008] added to which, p73 transcripts were independently shown to decline with age in human oocytes [Steuerwald et al., 2007]. The relevance of this work to the SAC is that the same experimenters subsequently demonstrated that TAp73 is important for SAC integrity by virtue of being required for proper BUBR1 localisation [Tomasini et al., 2009]. Intriguingly, sub-cellular protein mis-localisation compromised BUBR1 (and SAC) function although total oocyte BUBR1 levels were demonstrably increased [Tomasini et al., 2009]. Herein lies an important lesson: reduced expression of SAC proteins is not the only road to SAC compromise.
Measures of SAC Activity during Natural Reproductive Aging
The foregoing illustrates that mice engineered to carry defects in SAC function experience augmented aneuploidy rates and suffer reproductive compromise. However, the most important and physiologically relevant issue pertains to whether age-related SAC dysfunction exerts any material effect within a naturally aging female population. To-date, the data addressing this all-important question are very limited and often indirect.
One burgeoning experimental approach alluded to previously estimates SAC transcript levels in oocytes in order to provide an indirect measure of SAC functionality. Differences in SAC transcript levels have often been observed between young and old human oocytes [Steuerwald et al., 2007] and mouse oocytes [Pan et al., 2008] and give impetus to the notion that progressive SAC dysfunction is an accompaniment of aging.
Although the value of transcriptome mining strategies is undeniable, it is important that some of their limitations be considered. One of the caveats of gene profiling studies is that mRNA levels, not protein levels, are estimated. Furthermore, this is often undertaken at a solitary point in oocyte maturation, most frequently at the metaphase II-arrested stage. It is well established that stored maternal mRNAs are actively turned over during oocyte maturation [Gosden and Lee, 2010] so that levels at the meiosis II stage may not reflect those during meiosis I, the most vulnerable stage for chromosome mis-segregation. Furthermore, in vertebrate oocytes, protein translation is a dynamic event under stringent post-transcriptional regulation [Gosden and Lee, 2010] making it dangerous to assume that protein levels (the true determinants of functionality) will necessarily mirror mRNA levels. The available evidence indicates that mRNA levels can act as an indirect readout of protein in some, but not all, instances. Thus, on the one hand, Pan and colleagues [2008] sought to validate their microarray data using immunofluorescence for estimating the expression of 2 candidate proteins, ATRX and BRCA1, and found that reduced transcript abundance was indeed correlated with reduced protein expression. On the other hand, the above-mentioned study involving TAp73–/– mutant mice vividly highlights an mRNA/protein mismatch as increases in mRNA encoding the p73 isoform, ΔNp73, were not paralleled by increases in ΔNp73 protein levels [Tomasini et al., 2008].
A more direct approach for evaluating SAC activity during natural aging was recently undertaken and tested the prediction that there would be measurable differences between young and old mouse oocytes regarding readouts of SAC functionality [Duncan et al., 2009]. Experiments were based on a proven aging model as the mouse strain used was the same as had previously been shown to undergo a dramatic rise in aneuploidy with advancing maternal age [Pan et al., 2008]. The authors hypothesised that if SAC compromise occurs with aging then older oocytes should demonstrate 2 prominent phenotypes oft-observed in experimentally induced SAC-deficient oocytes: accelerated meiosis I transit [Homer et al., 2005c; McGuinness et al., 2009] and an inability to either delay or arrest meiosis I when challenged with spindle poisons [Wassmann et al., 2003; Homer et al., 2005b; Niault et al., 2007; McGuinness et al., 2009]. It was found that aged oocytes did not suffer the predicted downstream consequences of SAC compromise insofar as meiosis I was not accelerated and such oocytes retained the capacity to maintain an arrest in nocodazole [Duncan et al., 2009]. A prominent strength of this study was that the timing of anaphase I-onset could be correlated with the occurrence of aneuploidy in the same oocyte; oocytes which suffered aneuploidy did not experience accelerated transit through meiosis I. Therefore, on the basis of this, it was concluded that progressive SAC dysfunction is not a major contributor to maternal age-related aneuploidy.
Intriguingly, this group had previously reported age-related deregulation of SAC transcripts in the same strain of mice [Pan et al., 2008]. Can these seemingly discordant findings between gene profiling data (if indeed mRNA levels reliably forecast SAC protein levels) [Pan et al., 2008] and apparent SAC functionality [Duncan et al., 2009] be reconciled? One crucially important point to note is that targeted gene silencing strategies, either at the oocyte- or at the animal-level, aim to disrupt a single gene. In contrast, with aging, a raft of genes becomes deregulated; about 100 transcripts show greater than 2-fold variation between young and old eggs [Hamatani et al., 2004; Grøndahl et al., 2010]. Thus, although SAC components appear to succumb to aging, the net effect produced when additional genes (some of which are yet to be ascribed functions in oocytes) are also compromised is wholly unknown. For this reason, there are potential pitfalls when attempting to extrapolate a phenotype observed in a pure SAC mutant generated through single-gene silencing strategies to a naturally aging model in which multiple genes are likely to be simultaneously deregulated.
Taking this line of reasoning one step further so as to examine the possible consequences of additive effects, we can speculate on a hypothetical scenario that could plausibly arise with aging. Apart from core SAC components, another salient transcript observed to decline with age, on the basis of both microarray data and real-time quantitative RT-PCR, was that encoded by the SAC-related gene, CENPE (CENtromere Protien-E) [Pan et al., 2008]. CENP-E is a plus-end directed motor that is bifunctional in SAC signalling; on the one hand, CENP-E is a critical mediator of kinetochore-microtubule attachments and hence proper chromosome alignment while on the other hand, it is indispensable for amplifying the SAC signal and consequently for robust SAC-mediated arrest [Weaver et al., 2003]. SAC signal amplification is likely to be particularly important for effective SAC inhibition in the oocyte due to the oocyte’s very large volume (the volume of a mouse oocyte is ∼270 pl compared to only ∼6 pl for a typical somatic cell). In aged oocytes, impaired kinetochore-microtubule attachments arising from defective CENP-E function would be expected to invoke an SAC response. Yet, at the same time, CENP-E compromise would diminish maximal SAC signal generation, further exacerbating defects of core SAC components. Taken together therefore, aged oocytes could conceivably initiate an SAC-mediated delay in response to inherently flawed kinetochore-microtubule attachments. However, this delay would be unsustainable due to reduced levels of both core SAC components and ancillary inputs from CENP-E important for augmenting the SAC signal throughout the voluminous oocyte. Under such circumstances, after an initial delay, the SAC signal would prematurely dissipate and, in spite of the continued presence of misaligned chromosomes, oocytes would ultimately progress to metaphase II, thereby exacting a high aneuploidy toll. Thus, this is one conceivable scenario in which meiosis I transit might not be accelerated as hypothesised by Duncan et al. [2009] (and indeed, could even be delayed) in spite of SAC compromise. Such a scenario may not be far wide of the mark as older oocytes were highly aneuploid and showed a trend towards either delayed or arrested progression through meiosis I [Duncan et al., 2009]. Furthermore, aged human oocytes are well known to exhibit multiple misaligned chromosomes [Battaglia et al., 1996], a phenotype consistent with that predicted of faulty CENP-E functionality.
Concluding Remarks and Future Directions
The cellular circuitry required to execute and monitor chromosome segregation during mitosis in somatic cells has been worked out in considerable detail and has been an invaluable template for guiding investigation into meiosis I regulation. However, it is becoming increasingly apparent that SAC signalling in mammalian oocytes, whilst bearing many similarities with the mitotic SAC, does not exactly replicate it [Homer et al., 2005a]. Some prominent facets of the mammalian oocyte not shared by mitosis must be built into equations that aim to calculate SAC transduction events, including acentrosomal spindle assembly, the unique schedule of APC/C activities and the large oocyte volume.
Since the kinetochore is a critical nexus in SAC signalling, it will be important to identify key proteins required for forming kinetochore-microtubule attachments in mammalian oocytes and examine how they feed into the SAC cascade. Although microtubule motors such as CENP-E are likely to play important roles in this regard, exactly how they impose themselves on chromosome alignment when spindles assemble in the absence of centrosomes is not known. More also needs to be known about APC/C regulation in oocytes, such as the mechanism by which BUBR1 modulates CDH1 stability and whether there could be mammalian meiosis-specific APC/C co-activators akin to AMA1 in S. cerevesiae and cortex in Drosophila[see Pesin and Orr-Weaver, 2008].
With the explosion of gene expression profiling, it is imperative that we gain more insight into post-transcriptional regulation of SAC genes in mammalian oocytes so that the significance of observed alterations in mRNA levels can be better interpreted. Based on protein estimation by Western blotting, it is clear that MAD2 and BUBR1 undergo net synthesis during progression through meiosis I [Homer et al., 2005c, 2009]. Given that oocytes are transcriptionally quiescent following GVBD, such increases in MAD2 and BUBR1 suggest that SAC components are subject to considerable post-transcriptional control. Defining the nature of this regulation will be informative as declining levels of any such regulators, and not just SAC components themselves, could also heavily influence SAC protein abundance.
Due to the experimental limitations associated with mammalian oocyte research, addressing these all-important questions will be a formidable undertaking. Even more challenging but necessary will be the functional interrogation, albeit on a scaled-down level, of meiosis I control in human oocytes. With the trend towards delaying motherhood showing no real sign of relenting, exposing the secrets of the enigmatic oocyte is a foremost priority and will require that these challenges are met head-on with novel and innovative approaches.
Acknowledgements
This work is supported by a Wellcome Trust Clinical Fellowship (082587/Z/07/Z). I am grateful to members of the Carroll and Fitzharris labs.

