

Abstract
Angiogenesis is the formation of new blood vessels from pre-existing structures, and is a key step in tissue and organ development, wound healing and pathological events. Changes in cell shape orchestrated by the cytoskeleton are integral to accomplishing the various steps of angiogenesis, and an intact cytoskeleton is also critical for maintaining newly formed structures. This review focuses on how the 3 main cytoskeletal elements – microfilaments, microtubules, and intermediate filaments – regulate the formation and maintenance of angiogenic sprouts. Multiple classes of compounds target microtubules and microfilaments, revealing much about the role of actin and tubulin and their associated molecules in angiogenic sprout formation and maintenance. In contrast, intermediate filaments are much less studied, yet intriguing evidence suggests a vital, but unresolved, role in angiogenic sprouting. This review discusses evidence for regulatory molecules and pharmacological compounds that affect actin, microtubule and intermediate filament dynamics to alter various steps of angiogenesis, including endothelial sprout formation and maintenance.
Introduction
Angiogenesis is defined as new blood vessel growth from previously existing structures and occurs naturally in the placenta and uterus during pregnancy, in the ovary during follicle development, ovulation and corpus luteum formation, and during wound healing in healthy adults. In pathological conditions, uncontrolled angiogenesis can occur, resulting in various diseases and cancer [1,2]. Under quiescent conditions, endothelial cells form a protective barrier and line the vasculature. The endothelium and the protective barrier normally present must adapt rapidly to accommodate angiogenic sprout formation, where endothelial cells migrate from a quiescent monolayer with stable junctions and form new blood vessels in response to extravascular pro-angiogenic cues. Angiogenesis is a multi-step process which involves endothelial cell activation, degradation of the basement membrane, invasion, proliferation, lumen formation and stabilization. This process is regulated by a balance of pro- and anti-angiogenic molecules. Several key factors include, but are not limited to, growth factors [3,4,5,6,7,8,9], bioactive lipids [10,11,12,13], integrins [14,15,16,17,18,19,20,21], junctional proteins [22,23,24] and transmembrane proteinases [25,26,27,28,29]. These molecules ultimately transduce intracellular signals to the cytoskeleton, which orchestrates the various steps in angiogenesis.
Actin
Actin is a well-studied cytoskeletal element for controlling angiogenesis. An intact actin cytoskeleton is integral for cell motility and membrane protrusion events, which are key for mounting successful angiogenic responses. The actin cytoskeleton is tethered to the plasma membrane through adherens junctions and focal adhesions at cell-cell and cell-matrix contacts, respectively. As discussed in the sections below, inhibition of integrins, focal adhesions and cadherins [30,31,32,33,34] blocks angiogenic responses, indicating actin anchoring at these sites is critical to this process. Given the substantial evidence linking the above-described molecules associated with actin, it follows that the actin network and its associated proteins would be viable targets for anti-angiogenic therapies [for a review, see [35]]. The reader is also referred to other extensive reviews on migration [36], integrins [37], junctional regulation [22,38] and endothelial lumen formation [39] for further reading.
Cell-Matrix Interactions
Cell recognition and response to the extracellular matrix (ECM) is critical for successful sprouting responses. Blockade of various integrin subunits prevents angiogenic responses in multiple systems [14,21,40,41,42,43]. Integrin ligation results in activation of focal adhesion kinase, Ras, phosphoinositide-3 kinase-Akt signaling, mitogen-activated protein kinases, Src and Rac, Rho, and Cdc42 GTPases [44,45,46,47], in addition to other molecules not listed. Furthermore, recognition of the ECM by some integrins (such as αvβ3) results in the phosphorylation of integrin cytoplasmic tyrosine residues, which in turn promotes recruitment of intracellular adaptor proteins [48,49]. Integrin knockout models suggest that functional compensation occurs between various integrin subunits, and that embryonic and adult vasculogenic and angiogenic events are controlled by distinct integrin subunits [50]. These data provide important insights into the ECM and integrins, which transduce downstream signals and promote angiogenesis in development and pathological situations.
Small GTPases
The Rho family of small GTPases are master regulators of the actin cytoskeleton [51] and have been shown to be involved in angiogenesis. GTPases are perfectly suited to regulate actin dynamics, which are key for cell motility, protrusion formation and establishing polarity. Cdc42 and Rac1 mediate endothelial lumen formation [52,53,54]. Fitting with these data, expression of Cdc42 and Rac1 improved the architecture of pathological angiogenesis stimulated by VEGF administration [55,56]. Davis and colleagues [57 ]have provided biochemical evidence to link Cdc42 to the α2β1 integrin, which is required for lumen formation and endothelial morphogenesis. These data explain how matrix recognition of a pro-morphogenic substrate (such as collagen type I [58]) can induce endothelial lumen formation and sprouting responses, namely by coupling matrix recognition (via α2β1) with cytoskeletal rearrangements (via Cdc42) and matrix proteolysis (via membrane type I matrix metalloproteinase, MT1-MMP) [57]. In addition, Cdc42 can form a polarity complex with atypical protein kinase C and protease activated receptor 3 (Par 3), which directs lumen formation and sprouting [59]. These data highlight that endothelial polarity is established during angiogenic responses, and that the Cdc42 and Rac1 GTPases are required for polarization.
In addition to Cdc42 and Rac1, the small GTPases Rho and Rap1 have been investigated in angiogenesis. Collagen type I activated Src kinase and Rho through β1-integrins [60], and Rho activation stimulated endothelial cell assembly into new blood vessels in a mouse skin model of angiogenesis [61]. Further evidence for an involvement of Rho is provided by a report that Syx, a synectin-binding guanine exchange factor for Rho, may interact with angiomotin, an endothelial junctional protein, to direct sprouting [62]. Separate studies have demonstrated that silencing of the cerebral cavernous malformation 2 (CCM2) gene in endothelial cells activates Rho and impairs endothelial junctions to block vacuole and lumen formation but maintain initial sprouting responses [63]. Expression of dominant negative isoforms of Rho and treatment with C3 exoenzyme alone do not significantly alter vacuole and lumen formation [53], suggesting the additional change in junctional permeability contributes to a defect in lumen formation. Interestingly, CCM2 forms an intracellular complex with CCM1 and CCM3. CCM1 is also known as KRIT-1 and forms a complex with the Rap1 GTPase, which is involved in regulating endothelial permeability and junctional integrity [64,65]. In separate studies, Ras-associated protein 1 (Rap1), a small GTPase which associates with both integrins and cadherins, is required for angiogenesis [66], and is essential for the conformational activation of β1-integrins and postnatal neovascularization in endothelial cells [67]. These studies emphasize important roles for small GTPases in mediating various steps of angiogenesis, and highlight that cross-talk between small GTPases and endothelial junctions is critical for successful angiogenic responses.
Junctional Proteins
The endothelium lines the inner surface of the vascular system and is arranged as a monolayer. Another class of molecules involved in cell-cell communication that is integral in vessel organization and sprouting responses is the transmembrane adhesion receptors that maintain the integrity of endothelial junctions. Interendothelial junctions contain three complex junctional structures – including adherens junctions, tight junctions and gap junctions. Gap junctions are communication structures which allow the passage of small-molecular-weight solutes between neighboring cells. Tight and adherens junctions are mainly responsible for intercellular adhesion via the formation of actin filament-associated protein complexes along transmembrane adhesion sites [22] and regulate the permeability and organization of blood vessels [23,24].
Vascular endothelial (VE)-cadherin is the major determinant of endothelial adherens junctions. In addition, regulation of VE-cadherin activity or its presence at cell contacts is essential for controlling vascular permeability. VE-cadherin, like other members of the cadherin family, is linked through its cytoplasmic tail to p120, β-catenin and plakoglobin. β-Catenin and plakoglobin bind to α-catenin, which interacts with several actin-binding proteins, including α-actinin, ajuba, zonula occludens-1 (ZO-1) and others, forming a large and dynamic protein complex at cell contacts [68]. Tyrosine phosphorylation of β-catenin reduces its affinity for the cadherin cytoplasmic tail and increases its turnover at junctions, thereby destabilizing adherens junctions [69,70]. Mice deficient for VE-cadherin die at midgestation of vascular malformations [71]. Defects are more severe in the extraembryonic placental vasculature. No capillary plexus is formed in the allantois, although electron microscopy shows that interendothelial junctions do form. Thus, VE-cadherin seems dispensable for initial vasculogenesis, but is required for subsequent remodeling and vascular morphogenesis [71].
In addition to molecules responsible for maintaining junctional integrity, the Notch pathway has been reported to modulate angiogenesis [72,73]. Endothelial cells express Notch1 and Notch4 surface receptors, along with their ligands, Jagged1 and Jagged2 (homologs of Drosophila serrate-like proteins) and Delta-like 1 (DLL1) and DLL4. Notch signaling is initiated when the extracellular domain of the receptor recognizes ligand expressed on a neighboring cell, which cleaves the Notch receptor and releases the extracellular domain. The intracellular domain is released and translocates to the nucleus to activate gene transcription [73]. Notch signaling regulates endothelial cell specification and the initiation of branching morphogenesis in multiple systems [74,75,76,77,78]. Interestingly, feedback may occur from cytoskeletal regulatory proteins to regulate Notch signaling. Cytoskeletal binding protein, CG3973, was recently identified as a transcriptional target of Notch signaling in Drosophila. CG3973 is a member of the Gas2-like family of proteins which have potential binding sites for both actin and microtubules. Roper and colleagues [79 ]have recently shown that CG3973 negatively regulates Notch signaling, because adult flies lacking CG3973 have higher levels of Notch activation in various tissues. Perhaps more interesting, new evidence is emerging that Notch ligands cross-talk with cell adhesion and cytoskeletal machinery. The cytoplasmic tail of Jagged-1 contains a PDZ recognition motif that interacts with afadin, a molecule associated with adherens junctions [80]. These data suggest an additional connection to the cytoskeleton independent of an ability to activate Notch signaling. Altogether, these recent publications suggest an increased level of sophistication, where the cytoskeleton is integrated with Notch signaling. It remains to be demonstrated whether such cross-talk with the endothelial cytoskeleton and Notch signaling pathways occurs.
Role for Proteins That Regulate Membrane-Cytoskeletal Interactions
Actin accessory and membrane scaffold proteins comprise an additional class of molecules that can regulate angiogenic responses. Three prominent actin-associated proteins are filamin, annexin 2 and moesin. Filamin isoforms A, B and C stabilize and bridge F-actin networks to the plasma membrane through membrane receptors or ion channels [81]. In addition, filamins bind a diverse array of cellular proteins, some of which include the small GTPases Rho, Rac, Cdc42 and RalA, allowing assembly of signaling complexes in various systems [82,83]. In endothelial cells, gene silencing of Filamin B increased focal adhesion assembly, reduced VEGF-induced migration and cord formation, and increased p21 activated kinase (PAK) activation; further, Filamin B formed a complex with Vav-2 and Rac1 [84]. These data stress a role for filamin in controlling endothelial migration and tube formation.
Annexin 2 is a multifunctional membrane scaffold protein that has been implicated in the formation of new blood vessels [85]. Annexin 2 binds F-actin and spectrin [86] and is thought to organize the interface between the cytoplasm and plasma membrane by interacting with membrane phospholipids and actin filaments [87,88]. Gene silencing studies indicate a role for annexin 2 in regulating adherens junctions, actin dynamics and tight junctions [89,90,91,92]. We have shown that annexin 2 is required for endothelial sprouting responses, and it complexes with VE-cadherin following endothelial stimulation with sphingosine 1-phosphate [93], a potent pro- angiogenic factor [94]. Annexin 2 depletion reduced endothelial barrier integrity and significantly reduced Akt phosphorylation, which suggests annexin 2 may control endothelial morphogenesis through an adherens-junction-mediated pathway upstream of Akt [93]. These data indicate a key role for annexin 2 in regulating endothelial sprouting responses.
Ezrin, radixin and moesin proteins interact with transmembrane proteins and the cytoskeleton. This feature allows these proteins to organize specialized membrane domains critical for signaling complexes [95]. Interestingly, endothelial cells express predominantly moesin, while epithelial cells and hepatocytes predominantly express ezrin and radixin [96,97]. Moesin is required for increased endothelial permeability responses to various cues, because knockdown of moesin blocks increases in permeability in response to TNF-α and other stimuli [98,99]. Essner and colleagues [100] report that moesin1 is required for lumen formation in developing intersegmental vessels in the zebrafish embryo. Moesin knockdown ablated VE-cadherin-positive adherens junctions, but not tight junctions. These data suggest VE-cadherin and moesin1 cooperate to establish and maintain endothelial polarity [100]. In separate studies, mice lacking moesin had delayed lumen formation in the dorsal aorta and a decreased amount of F-actin beneath the apical surface of the endothelium, which is where lumen formation is initiated [101]. In addition, CD34-sialomucins and podocalyxin were implicated with moesin, as well [101]. These studies reinforce that establishment of endothelial polarity is critical for proper angiogenic responses.
Microtubules
Microtubules normally ebb and flow and exhibit dynamic instability and treadmilling. Microtubule-binding drugs and vascular-disrupting agents are widely used to interfere with the formation of angiogenic structures and stimulate their collapse, which is also defined as breakdown or regression of existing structures. A summary of the effects of compounds that affect microtubule organization which have been used to study various steps in angiogenesis is summarized in table 1. Microtubule-binding drugs and vascular-disrupting agents suppress the dynamics of microtubules without appreciably changing microtubule mass and are undergoing various clinical trials [102,103]. These agents have provided useful tools to demonstrate that tubulin polymerization and stabilization are required for formation and maintenance of angiogenic structures, respectively. Microtubule depolymerizing agents – such as ZD6126, AVE8062, combrestatin A4, CYT997, JG-03-14, TH-482, vinblastine and vinflunine – can destabilize existing vascular networks. Microtubule-depolymerizing agents can also block sprout formation and include colchicine, combrestatin A4, embellistatin, 2-methoxyestradiol (2-ME), spongistatin, tubulysin A and XRP44X. Microtubule-stabilizing compounds – including docetaxel, epothilone B, IDN 5390, laulimalide and paclitaxel – also block sprout or cord formation. Thus, microtubule networks must remain intact for angiogenic network maintenance and stabilization, but also maintain dynamic properties that are required for the initial formation of angiogenic structures. Interestingly, intracellular levels of these compounds can accumulate in endothelial cells approximately 5 times higher than other cells [104,105,106].

Consequences of Microtubule Alterations
Microtubules are critical for successful cell division, intracellular transport and signaling. Microtubule-associated proteins are often misregulated in cancer, making tubulin and microtubules viable targets for chemotherapy. Microtubule targeting compounds are among the most effective classes of chemotherapeutics to prolong survival in patients with metastatic disease [162,163]. Described below are various intracellular pathways that are perturbed following microtubule disruption and stabilization. These include alteration of microtubule plus- and minus-end molecules, hypoxia-inducible factor (HIF) and various other processes. Microtubule flux is governed by regulation of microtubule growth at plus-ends and anchoring at minus-ends. Minus-end anchoring of microtubules originates at the centrosome with the aid of multiple accessory proteins. Ninein is one such microtubule minus-end anchoring protein [164,165]. Ninein is phosphorylated as endothelial cells undergo tubulogenesis, and enhanced in tip cells in endothelial outgrowths from embryoid bodies embedded in 3-D collagen matrices [166]. Disruption of the plus-ends of microtubules similarly disrupts endothelial motility and tubulogenesis [158]. Low doses of vinflunine inhibited endothelial cell motility that correlated with EB-1 mislocalization at microtubule plus-ends, decreased microtubule targeting to adhesion sites, decreased adhesion site dynamics and the formation of stable stress fibers [157]. These results illustrate that disruption of both plus- and minus-ends of microtubules significantly alters sprouting responses, and cross-talk between microtubules, actin and adhesion complexes is critical to sprouting.
Microtubule stabilizing and destabilizing drugs inhibit HIF-1α accumulation by disrupting microtubule function [167]. Giannakakou and colleagues [168] first reported that microtubule disruption dysregulates HIF; 2ME2, taxol and vinblastine blocked nuclear accumulation of HIF-1α, VEGF secretion and angiogenic responses. In addition, nuclear HIF-2α accumulation and VEGF production were blocked in primary endothelial cells [168]. Similar results were observed with M410, which blocked cell cycle progression and induced microtubule depolymerization in a human colon carcinoma line [145]. Stathmin is a cytosolic protein that binds the α/β-tubulin dimer and depolymerizes microtubules. Stathmin knockdown in primary endothelial cells led to microtubule stabilization and inhibited HIF-1α protein accumulation and VEGF expression [169]. It appears that 2ME-2, Taxol, vinblastine, M410, ENMD-1198 and epothilone B all reduce HIF-1α expression, nuclear HIF-1α accumulation and VEGF expression in endothelial and tumor cells, and these alterations correlate to decreased angiogenic responses in vivo [145,167,168,170]. Thus, successful intranuclear transport of HIF appears to require the ability of microtubules to polymerize or depolymerize, as stathmin knockdown and Taxol treatment also block HIF-1α nuclear translocation [168,169]. This action has been linked to the ability of these compounds to target β-tubulin [167].
Cross-Talk between Microtubules and Actin
Communication between microtubules, actin and focal adhesions are critical for successful cell motility [171]. Microtubule-binding drugs can alter structures associated with actin assembly, such as adherens junctions and focal adhesions. Both microtubule stabilizing and destabilizing compounds can prevent endothelial focal adhesion formation and assembly [102,133,137,157]. Inhibiting microtubules can cause a loss of cell polarity and block formation of lamellipodia [172]. Consistent with this observation, non-toxic doses of docetaxel, epothilone B and vinblastine significantly inhibited endothelial cell migration, invasion and cord formation on matrigel, which was ascribed to reduced F-actin stress fiber formation, appearance of nuclear F-actin rings and early inhibition of Rac1 and Cdc42 activity [132]. Higher doses of vinblastine, a microtubule-disrupting agent, induced rapid collapse and apoptosis of established endothelial networks in 3-D collagen matrices [153]. Interestingly, inhibiting actin polymerization with cytochalasin D did not induce collapse of pre-formed 3-D endothelial networks [153]. The vinblastine-induced collapse required Rho GTPases and was blocked with C3 exoenzyme and dominant negative forms of RhoA and RhoC, indicating microtubule depolymerization induced Rho GTPase activation and collapse of endothelial networks [153]. In separate studies, Luis and colleagues [173] recently reported that MVL-PLA2, a phospholipase A2 from snake venom, increased microtubule dynamics and blocked angiogenesis. MVL-PLA2 likewise disrupted focal adhesions, contributing to failed angiogenic responses [173]. In line with this observation, TAE226 inhibition of focal adhesion kinase blocked tube formation in vitro and microvessel density in subcutaneous tumors [174]. These studies underscore the interplay between focal adhesions and dynamic microtubule networks, and illustrate the link between microtubule perturbation and actin regulatory molecules.
Newly forming endothelial cells exhibit a polarized phenotype to guide sprout formation in response to angiogenic stimuli [74,175,176]. Crews and colleagues [177] have recently reported the activation of disheveled associated activator of morphogenesis (DAAM1) interferes with endothelial cell division, migration and angiogenesis that correlated with microtubule stabilization. Davis and colleagues [57] have demonstrated that a polarity complex forms with Cdc42 that guides endothelial cell outgrowth. In line with this, disruption of microtubules may have the added ability to downregulate expression of VEGFR2, but not VEGFR1 [178]. This would be predicted to have a significant impact on angiogenic sprouting because VEGFR2 is enriched in polarized tip cells [77,179] and is vital for promoting angiogenic responses [180,181]. Altogether, these data suggest a complicated cross-talk exists between extracellular cues and establishment of cellular polarity, which is ultimately accomplished through rearrangement of cytoskeletal elements.
Intermediate Filaments
The intermediate filament cytoskeleton connects the plasma membrane to the nucleus and in many cases displays a tight interaction with the nuclear lamina and nuclear cytoskeleton. Intermediate filament proteins were classically thought to function solely for mechanical stabilization of cells, but are more recently being recognized as regulators of signal transduction events [182,183,184]. Because intermediate filaments are resistant to detergent solubilization, they were once predicted to be stationary; however, intermediate filaments engage in various movements that are linked with assembly, disassembly and subcellular organization [185]. Vimentin is a type III intermediate filament that is expressed in cells of mesenchymal origin, including endothelial cells [186]. Thus, vimentin will be the primary focus of the following discussion.
Vimentin intermediate filament networks are highly dynamic [187], and the polymerized filament is formed through the stacking of dimers and tetramers [188,189,190]. Each monomer contains a central α-helical rod domain that is flanked by an N-terminal head and C-terminal tail [191]. Vimentin polymer organization is dependent on the phosphorylation state, because the introduction of a negatively charged phosphate group, predominantly in the head region, frees soluble vimentin from the polymerized network [192,193,194,195,196]. Phosphorylation may reduce the positive charge in the head and tail domain and reduce the propensity for vimentin polymerization [197,198]. In addition, the N-terminal head domain is cleaved by calpain [199,200]. Fragments of vimentin produced by calpain cleavage do not assemble into intermediate filaments [201], and injection of N-terminal vimentin peptides collapsed intermediate filament networks [202]. These studies show that both phosphorylation and cleavage disrupts vimentin polymerization, producing soluble vimentin, which has been previously suggested to function as an intracellular signal transducer [182].
The underlying cell processes controlled by intermediate filaments remain obscure and have not been investigated in detail in endothelial cells or with respect to blood vessel formation, although many studies have revealed important functions for vimentin in smooth muscle cells, fibroblasts and tumor cells. Mild phenotypic alterations were observed in the original report of vimentin-null animals [203], but subsequent studies have identified defects in endothelial barrier function [204] and fibroblast contraction during wound healing [205]. Vimentin-null mice also have cerebellar defects, impaired motor coordination, and purkinje cell necrosis [206]. Of interest here, Eckes et al. [205] reported a lag in granulation tissue formation that has not been investigated further. This anecdotal evidence supports the possibility that vimentin regulates angiogenic sprouting during granulation tissue formation. Santilman et al. [207] have recently reported that vimentin binds caveolin (Tyr14) and co-localized with caveolin at the anterior portion of cells extending protrusions through Boyden chambers. Caveolin-1 polarization in 3-D migrating endothelial cells required the phosphorylatable Tyr14 residue of caveolin-1. Immunoelectron microscopy further indicated that caveolin-1 was distributed along cytoskeletal structures in the anterior of transmigrating endothelial cells [208]. A similar polarization of vimentin and Tyr14 caveolin was reported during retinal development in vivo [207], although vimentin fibers seen in 2-D cultures were reported in endothelial cells extending protrusions across Boyden chamber membranes [207]. Vimentin-null mice have reduced corneal neovascularization [209] and hypoxia-induced retinal neovascularization [210]. Whether these effects can be explained by direct defects in endothelial function remains to be demonstrated.
While classically thought to provide mechanical support to cells and tissues, emerging evidence suggests additional roles for intermediate filaments. Peptide-induced disassembly of vimentin intermediate filaments dramatically altered fibroblast shape as well as microfilament and microtubule organization [202], yet, in mice lacking vimentin, cytoskeletal organization is normal [203]. These seemingly conflicting observations suggest that vimentin monomers are signaling intermediates [186]. The mechanical properties of intermediate filaments in vitro and the fact that in most cells they form interacting networks between the cell surface and the nucleus supports the hypothesis that the intermediate filament cytoskeleton provides a scaffold to sense and transduce mechanical signals. For example, applying 12 dynes/cm2 shear stress to vascular endothelial cells rapidly displaced the extensive 3-D networks of vimentin intermediate filament networks at the apical surface. Intermediate filaments are displaced approximately 1 µm in a 3-min time period. While these movements occurred throughout the cytoplasm, there was relatively more translocation at the apical surface than the basal surface of the cell [211]. The idea that vimentin may act as a mechanotransducer is supported by defective flow-induced vasodilation responses in vimentin-null animals [212]. Recent work by Fainzilber and colleagues [213,214] also highlights the ability of calpain-cleaved vimentin fragments to act as intracellular chaperones to alter gene transcription following crush injuries. Newly synthesized vimentin was cleaved by calpain and bound to p42 and p44 mitogen-activated protein kinases (pErk1/2) [213], protecting phosphorylated Erks from dephosphorylation [214] while also transporting them from distal sites of injury to the nucleus to drive transcription. Injured neurons from vimentin-null mice could not perform pErk1/2 transport, and this effect was correlated with delayed recovery of sensation [213]. Thus, soluble non-polymerized vimentin fragments transduced intracellular signals by chaperoning activated Erk1/2 in regenerating neurons. Finally, treatments that activate signal transduction pathways can change intermediate filament distribution [215]. Given the ability of vimentin to function in signal transduction, it is reasonable to predict that vimentin is involved in initiating angiogenic sprouting responses, although this has not been demonstrated definitively to date. One limitation to thoroughly investigating intermediate filaments is a lack of pharmaceutical compounds that specifically target intermediate filament networks. Withaferin A has been reported to bind vimentin dimers and stabilize vimentin networks [209], and withaferin A has been reported to block tumor angiogenesis [209,216,217]. Although the above-described studies suggest an important function for vimentin in angiogenesis, none to date have defined a mechanism for vimentin regulation of angiogenic network formation. Pilot studies in our laboratory reveal that silencing vimentin expression in primary endothelial cells significantly impairs endothelial sprouting into three-dimensional collagen matrices (data not shown). Further investigation will be required to demonstrate a functional role for the intermediate filament vimentin in orchestrating angiogenic responses and the mechanism through which this occurs.
Cross-Talk between Vimentin and Other Cytoskeletal Elements
Vimentin is an abundantly expressed protein that associates with various intracellular molecules. Vimentin intermediate filaments are associated with the centrosome [218] and bind activated Rac and Cdc42 [219], phospholipase A2 [220], and the molecular motors kinesin, dynein and dynactin [221,222]. Vimentin is important for lipoprotein-derived cholesterol esterification [223]. Interestingly, various protein kinase C isoforms associate with vimentin intermediate filaments [224,225,226], and PKC-mediated vimentin phosphorylation regulates recycling of the β1-integrin subunit during cell migration [227]. Plectin and intermediate filament associated protein-300 bind and cross-link vimentin to microtubules, microfilaments and membrane adhesion complexes [228,229,230]. Vimentin associates with filamin A [231], α-crystallin [232], cGMP kinase [233] and Yes kinase [234]. The carboxy terminus of vimentin binds actin [235], and detyrosinated tubulin recruits vimentin intermediate filaments to microtubules [236]. Lastly, deleterious effects, including cataracts and lens degeneration, are seen in mice that overexpress vimentin [237]. These data underscore the intricate relationships established by molecules to integrate actin and microtubule dynamics with the vimentin intermediate filament cytoskeleton. Further work will be required to continue to tease apart how the various cytoskeletal moieties interact with one another to control motility and angiogenic sprouting.
Intracellular Localization of Actin, Tubulin and Vimentin during Endothelial Sprouting
We investigated the distribution of microtubules, vimentin and actin in 2-D versus 3-D endothelial cultures using indirect immunofluorescence (fig. 1). Filamentous networks for α-tubulin, vimentin and α-, β-, γ-actin were seen in 2-D endothelial monolayers that extended from nucleus to plasma membrane (fig. 1, upper panels). The arrangement of these networks was distinct, however, for endothelial cells invading 3-D collagen matrices, which mimics angiogenic sprout initiation. Large highly aligned bundles in 3-D cultures were observed for α-tubulin and vimentin that were maintained continuously throughout extensions of peripheral processes. These distinctions are depicted in the schematic in figure 2. The images highlight key differences in cytoskeletal arrangement in 2-D monolayers (those forming stable junctions) and 3-D invading endothelial cells, which form sprouting structures.
![Fig. 1. Immunofluorescence analysis of tubulin, vimentin and actin arrangement in 2-D versus 3-D cultures. Top row: Human umbilical vein endothelial cells (passage 5) were seeded on 18-mm circular coverslips coated with collagen type I (20 µg/ml) and allowed to attach overnight. Cells were treated with 1 µM S1P for 1 h before being fixed and processed for immunofluorescence as described in the ‘Appendix’. Primary antibodies utilized were anti-α-tubulin (clone DMA1; Sigma-Aldrich; 1:100), anti-vimentin [V9 FL (p) epitope; Santa Cruz; 1:100] and anti-actin (AB-1 clone JLA20; EMD Biosciences; 1:25). Images were collected with a Nikon Eclipse TE2000-U microscope. ×60. Bottom row: Cells were seeded on 3-D collagen type I matrices (2.5 mg/ml) with growth factors and 1 µM S1P for 24 h, fixed in 4% paraformaldehyde in PBS for 30 min and processed for immunofluorescence. Images collected with a Zeiss Imager.A1m confocal microscope (0.5- to 1.5-µm stacks that were layered into 1 image). ×40. Arrowheads indicate extended peripheral processes.](https://karger.silverchair-cdn.com/karger/content_public/journal/jvr/48/5/10.1159_000324751/2/m_000324751_f01.jpeg?Expires=1716326361&Signature=3N5JfkGEbG6p5QIuLuHrA6QpBcpToyIlyXTmDzR6nn0reNKhPlXaDx1CApGpHae4kqijwFWDq~EPheKMHAApD~QfyaeaJz4573ifRlzHcYR-15FzyiFpc0cocMN2YCICTfQFMJ6c1B2H1KM3lo6nraG-o1RcIGUzFcGoAjIkZaXxoD34KETu4OCzIl44V2hz6hohcyItsVYvtJaF2VoHYJh8QniISlpE7Z4ubC~TJ4a2rxq7MJa5C83MN2vlVz02C3p9xGSu9qr88RIpNm-4a-jTxVmZQIdqBeNUjIFtCRkXwZXmcb75r8IX3dgQ2FGBAmIILG8~FNBU17L2Z8MQVg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Immunofluorescence analysis of tubulin, vimentin and actin arrangement in 2-D versus 3-D cultures. Top row: Human umbilical vein endothelial cells (passage 5) were seeded on 18-mm circular coverslips coated with collagen type I (20 µg/ml) and allowed to attach overnight. Cells were treated with 1 µM S1P for 1 h before being fixed and processed for immunofluorescence as described in the ‘Appendix’. Primary antibodies utilized were anti-α-tubulin (clone DMA1; Sigma-Aldrich; 1:100), anti-vimentin [V9 FL (p) epitope; Santa Cruz; 1:100] and anti-actin (AB-1 clone JLA20; EMD Biosciences; 1:25). Images were collected with a Nikon Eclipse TE2000-U microscope. ×60. Bottom row: Cells were seeded on 3-D collagen type I matrices (2.5 mg/ml) with growth factors and 1 µM S1P for 24 h, fixed in 4% paraformaldehyde in PBS for 30 min and processed for immunofluorescence. Images collected with a Zeiss Imager.A1m confocal microscope (0.5- to 1.5-µm stacks that were layered into 1 image). ×40. Arrowheads indicate extended peripheral processes.
Immunofluorescence analysis of tubulin, vimentin and actin arrangement in 2-D versus 3-D cultures. Top row: Human umbilical vein endothelial cells (passage 5) were seeded on 18-mm circular coverslips coated with collagen type I (20 µg/ml) and allowed to attach overnight. Cells were treated with 1 µM S1P for 1 h before being fixed and processed for immunofluorescence as described in the ‘Appendix’. Primary antibodies utilized were anti-α-tubulin (clone DMA1; Sigma-Aldrich; 1:100), anti-vimentin [V9 FL (p) epitope; Santa Cruz; 1:100] and anti-actin (AB-1 clone JLA20; EMD Biosciences; 1:25). Images were collected with a Nikon Eclipse TE2000-U microscope. ×60. Bottom row: Cells were seeded on 3-D collagen type I matrices (2.5 mg/ml) with growth factors and 1 µM S1P for 24 h, fixed in 4% paraformaldehyde in PBS for 30 min and processed for immunofluorescence. Images collected with a Zeiss Imager.A1m confocal microscope (0.5- to 1.5-µm stacks that were layered into 1 image). ×40. Arrowheads indicate extended peripheral processes.
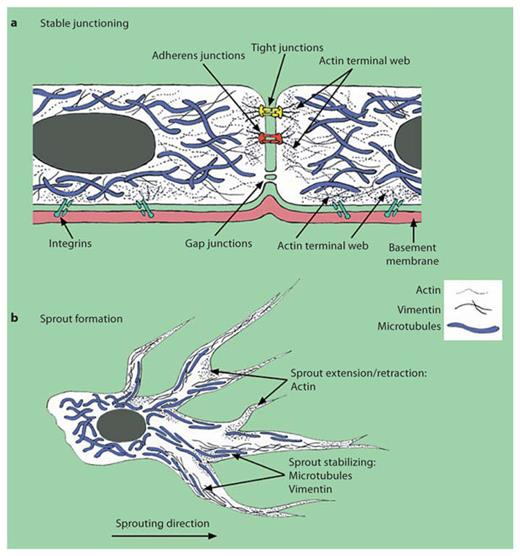
Schematic depicting microtubule, vimentin and actin localization in quiescent versus activated endothelial cells: quiescent endothelial cells (a) and sprouting endothelial cells (b). Placement is based on the data shown in figure 1.
Schematic depicting microtubule, vimentin and actin localization in quiescent versus activated endothelial cells: quiescent endothelial cells (a) and sprouting endothelial cells (b). Placement is based on the data shown in figure 1.
Microtubules are clearly required for maintenance of both 2-D monolayers and endothelial sprouting structures. Treatment with either microtubule-destabilizing or -stabilizing compounds collapses existing structures ( [130,142,153] and data not shown). Actin arrangement in 3-D, however, was punctuate and distinctive from 2-D arrangements. Actin foci observed in 3-D are established at critical subcellular locations (e.g. strategic branch points). It is possible that fine extended protrusions are more susceptible to collapse because of differences in adherence to the ECM. We have reported that endothelial cells continually extend and retract peripheral processes while advancing through 3-D collagen matrices, and this extension and retraction required a disintegrin and metalloproteinase 17 (ADAM17) [238]. These extended peripheral processes (indicated by arrowheads in fig. 1) often extend around individual collagen fibers, but do not alter collagen density or arrangement significantly [239]. At the same time, extension and retraction of peripheral processes is occurring at the leading edge of the sprout, while individual tunnels are generated by trailing endothelial cells (analogous to stalk cells) which generate the lumen portion of the structure [239,240,241]. The various states initiated in angiogenesis are depicted in figure 3, where quiescent endothelial cells with stable junctions are activated by release of pro-angiogenic stimuli. This endothelial activation triggers sprout initiation, endothelial invasion and new blood vessel formation.
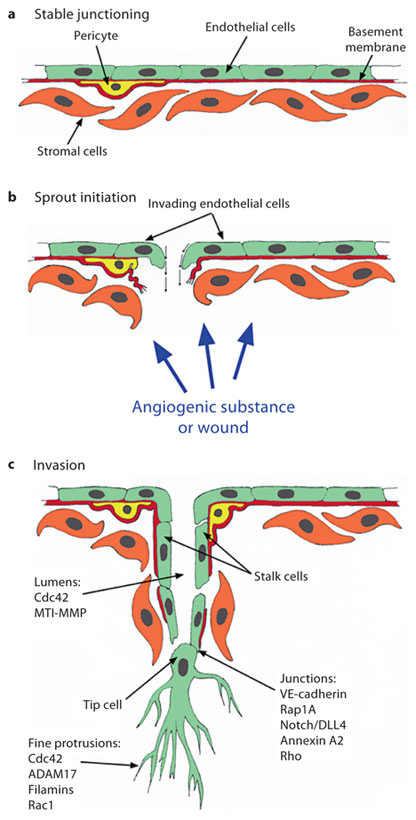
Schematic illustration illustrating key steps in angiogenesis, along with molecules that transduce signals to the cytoskeleton to induce sprouting angiogenesis. a Quiescent endothelium exhibiting intact basement membrane (red), mural cell (yellow) and intact junctions. b Sprout initiation is stimulated by local production of angiogenic factors, which disrupt junctions and basement membrane integrity to initiate sprouting responses. c Sprout extension and new vessel growth. Key molecules that control lumen formation, junctional signaling and fine protrusion formation are indicated.
Schematic illustration illustrating key steps in angiogenesis, along with molecules that transduce signals to the cytoskeleton to induce sprouting angiogenesis. a Quiescent endothelium exhibiting intact basement membrane (red), mural cell (yellow) and intact junctions. b Sprout initiation is stimulated by local production of angiogenic factors, which disrupt junctions and basement membrane integrity to initiate sprouting responses. c Sprout extension and new vessel growth. Key molecules that control lumen formation, junctional signaling and fine protrusion formation are indicated.
It appears that cytoskeletal arrangements within endothelial cells extending sprouts into an intact 3-D ECM versus endothelial cells lining and forming a monolayer are distinct, and cellular responses to various cues rely heavily on both matrix integrity and endothelial cell polarization, which would be expected to be distinct at various segments of developing sprouts. Certainly, these interactions are dynamic, as developing microvascular structures alter the mechanical properties of the surrounding ECM with time [239,242]. The cell-matrix interactions between newly forming angiogenic structures are mediated through integrins [14,15,21,40,41,42,43,243,244], and maintaining endothelial attachments are dependent on integrin-mediated contacts, because integrin antagonism collapses sprouting structures [21]. In addition, varying the matrix density, which presumably alters the availability of integrin binding sites within the ECM, alters sprouting responses and lumen size in various assays ( [245,246] and our unpublished observations). Although extensive studies to date have revealed important clues, the precise molecular cues responsible for the intricate processes of initiating, propelling and maintaining endothelial sprouting structures remain to be definitively identified.
Altogether, the studies described here illustrate a key role for the cytoskeleton in angiogenic sprout initiation and maintenance, which are summarized in figure 3. Continued investigations will clarify how cross-talk between microtubule, microfilament and intermediate filament networks is accomplished for these vital processes. Also of interest is how various cytoskeletal networks integrate with and respond to the key signaling pathways known to regulate angiogenesis. Lastly, continued discovery of new compounds that target cytoskeletal components and normalize aberrant angiogenesis will provide additional tools to combat pathological angiogenesis in disease.
Appendix
Reagents
Human umbilical vein endothelial cells were purchased from Lonza and maintained as described [247].
Immunofluorescence Analyses
Endothelial cells seeded on coverslips were fixed in 2% paraformaldehyde in PBS for 10 min, rinsed twice with Tris-glycine buffer, permeabilized for 15 min with 0.5% Triton X-100 in PBS, and blocked overnight in blocking buffer (1% BSA, 0.2% sodium azide, 0.1% TX-100, 1% goat serum in TBS). Coverslips were washed for 5 min with wash buffer (0.1% Triton X-100 in PBS). Secondary antibodies (goat anti-mouse, Jackson Immuno Research; 1:60) in blocking buffer were added for 1 h at room temperature. Samples were washed for 5 min, rinsed in water and mounted. Endothelial cells invading 3-D collagen matrices were allowed to invade for 24 h, fixed in 4% paraformaldehyde in PBS for 30 min, and rinsed twice with Tris-glycine buffer. Samples were cut to generate a side view, permeabilized for 1 h (0.5% Triton X-100 in PBS), and blocked overnight in blocking buffer (1% BSA, 0.2% sodium azide, 0.1% TX-100, 1% goat serum in TBS). Primary antibody was added at room temperature for 3 h. Samples were washed for 45 min with wash buffer. Secondary antibodies (goat anti-mouse Alexa Fluor® 488; Molecular Probes) were diluted 1:300 in blocking buffer and incubated at room temperature for 1 h. Samples were washed overnight for the α-tubulin and vimentin. Samples stained with actin were washed for 30 min, rinsed in water, and mounted.
Acknowledgments
This work was supported by NIH R01HL09576, National Research Initiative Competitive Grants No. 2009-35203-05725 from the USDA National Institute of Food and Agriculture, and National Research Initiative Competitive Grant No. 2006-35203-17199 from the USDA Cooperative State Research, Education, and Extension Service.
We thank Dr. Jeffrey Essner for reading the manuscript and for helpful suggestions. We thank Adriana Mendoza for assistance with immunofluorescence studies.
