

Abstract
Down syndrome (DS; trisomy 21) is one of the most common genetic causes of intellectual disability, which is attributed to triplication of genes located on chromosome 21. Elevated levels of several microRNAs (miRNAs) located on chromosome 21 have been reported in human DS heart and brain tissues. The Ts65Dn mouse model is the most investigated DS model with a triplicated segment of mouse chromosome 16 harboring genes orthologous to those on human chromosome 21. Using ABI TaqMan miRNA arrays, we found a set of miRNAs that were significantly up- or downregulated in the Ts65Dn hippocampus compared to euploid controls. Furthermore, miR-155 and miR-802 showed significant overexpression in the Ts65Dn hippocampus, thereby confirming results of previous studies. Interestingly, miR-155 and miR-802 were also overexpressed in the Ts65Dn whole blood but not in lung tissue. We also found overexpression of the miR-155 precursors, pri- and pre-miR-155 derived from the miR-155 host gene, known as B cell integration cluster, suggesting enhanced biogenesis of miR-155. Bioinformatic analysis revealed that neurodevelopment, differentiation of neuroglia, apoptosis, cell cycle, and signaling pathways including ERK/MAPK, protein kinase C, phosphatidylinositol 3-kinase, m-TOR and calcium signaling are likely targets of these miRNAs. We selected some of these potential gene targets and found downregulation of mRNA encoding Ship1, Mecp2 and Ezh2 in Ts65Dn hippocampus. Interestingly, the miR-155 target gene Ship1 (inositol phosphatase) was also downregulated in Ts65Dn whole blood but not in lung tissue. Our findings provide insights into miRNA-mediated gene regulation in Ts65Dn mice and their potential contribution to impaired hippocampal synaptic plasticity and neurogenesis, as well as hemopoietic abnormalities observed in DS.
Introduction
Down syndrome (DS) is caused by the triplication of chromosome 21 of mainly maternal origin in humans [1,2,3]. It is a common noninheritable genetic disorder, occurring in approximately 1 out of every 590 live births worldwide (calculated based on recently published multinational epidemiological results [4]). Some of the most prominent features of the DS phenotype include mental retardation as well as an increased incidence of congenital heart disease, hypothyroidism and leukemia [5,6,7]. In addition, DS infants are at increased risk for the development of persistent pulmonary hypertension [8,9]. Gene dosage is believed to play a significant role in determining the DS phenotype [5,7,10,11]. Indeed, some DS mouse model studies have shown a 1.5-fold increase in the expression of many genes located on the triplicated gene segment [12,13,14,15]. However, a significant number of transcripts and proteins encoded by human chromosome 21 and nonchromosome 21 orthologous genes do not follow gene dosage ratios [12,13,14,15]. While gene dosage may contribute to the phenotype associated with DS, an exact mechanism and specific gene networks underlying DS abnormalities have yet to be elucidated [16,17,18].
No single gene or region of human chromosome 21 has been found that is responsible for all the common features of DS [7,19]. Therefore, it would be expected that multiple genes and other factors working in concert are responsible for the major DS phenotypes. It is well established that the expression of different alleles, interactions between various genes, and the additive effects of altered expression over time contribute to the presence or severity of any given phenotype, which is likely the case in DS [7,20,21,22].
More recent studies have begun to focus on epigenetic alterations for variations in gene expression resulting in abnormal phenotype, disease process, or specific biomarker [23,24,25,26,27]. One specific area of interest has been the study of microRNAs (miRNAs) in neurodevelopmental disorders [28,29] such as DS [30,31,32]. miRNAs are single-stranded endogenous noncoding ∼22 nucleotide RNAs which regulate gene expression by destabilizing target mRNA and inhibiting protein translation [33,34]. However, several miRNAs have been recently implicated in upregulation of protein expression [35,36].
miRNAs are important regulators of developmental and evolutionary processes and play a key role in the pathogenesis of some acquired disorders [37,38]. Changes in miRNA expression can cause tumor suppression, activate oncogenes and/or accelerate tumorigenesis [39,40,41,42]. miRNAs are active within the CNS as well, where they regulate neuroplasticity through mediators such as cAMP responsive element binding protein (CREB1), methyl CpG binding protein 2 (MeCP2) and FMRP [43,44,45,46]. miRNAs are also found within the dendrites where they regulate local protein translation [47,48]. Therefore, miRNAs, if altered in DS, could impact synaptic function in the dendrites and contribute to the abnormal physiological properties of DS neurons [30,31,32]. We hypothesize that overexpression/dysregulation of a group of miRNAs, some possibly secondary to a gene dosage effect, may contribute to the pathology associated with DS through dysregulation of protein expression throughout development, in the adult brain, and during hematopoiesis.
Interestingly, a recent study has shown that human chromosome 21 harbors the sequence for five miRNAs (miR-99a, miR-125b-2, miR-155, miR-802 and let7-c) and that these miRNAs are overexpressed in DS human fetal brain and cardiac tissue [30,31,32]. Each miRNA located on chromosome 21 has multiple potential targets, though the exact targets and pathways affected have not yet been determined. In the presence of overexpressed miR-155 and miR-802, the resulting gene targets, including MeCP2, appear to be downregulated in both DS and Ts65Dn brain tissue [31,32]. MeCP2 and its target CREB are believed to play an important role in memory and learning and are highly expressed in the hippocampus [49,50]. SHIP1 [SH2 (Src homology 2)-containing inositol phosphatase-1] gene is predominantly expressed in hemopoietic cells and has been recently identified as a target of miR-155 [51,52,98]. SHIP1 is a member of inositol polyphosphate 5-phosphatases with some members such as SHIP2 and synaptojanin 1 (Synj1 overexpressed in Ts65Dn brain [53]). SHIP2 and synaptojanin 1 are known to play an important role in neuronal function and astrogliogenesis [54,55,56]. The only report that describes a neuronal role of SHIP1 relates it to axonal growth in cortical neurons via interactions with neogenin and uncoordinated Unc5H2 [57]. Outside the nervous system, we recently reported that miR-155 specifically reduced levels of SHIP1, thereby promoting phosphatidylinositol 3-kinase (PI3K)/Akt activation in cystic fibrosis lung epithelial cells [51]. In diffuse large B cell lymphoma, elevated levels of miR-155 and consequently diminished SHIP1 expression were triggered by stimulation of the proinflammatory cytokine tumor necrosis factor alpha [52]. While overexpression of miR-155 and miR-802 in DS and Ts65Dn have been reported, their exact contribution to the pathology and expression levels of other miRNA and mRNA in DS remains to be investigated.
The development of trisomy mouse models has opened a new window to unravel the molecular and developmental defects associated with abnormal chromosome number. The distal portion of mouse chromosome 16 is homologous to nearly the entire long-arm of human chromosome 21. Mice with segmental trisomy 16 (Ts65Dn) are considered the best-characterized animal model of DS (fig. 1). The portion of chromosome 16 (MMU 16) that is triplicated in the Ts65Dn model contains approximately 99 of the 170 orthologous genes present on human chromosome 21 (HSA21) [58,59]. This model survives birth and lives into adulthood allowing for studies of the pre- and postnatal effects of trisomy [60,61]. One of the significant defects observed in the Ts65Dn mouse is an impairment in spatial learning tasks [62,63,64] indicating a hippocampal deficit. The hippocampus plays a critical role in learning and memory and is a site for long-term synaptic plasticity. Neuropsychological evidence suggests that hippocampal dysfunction is also present in adolescents and young adults with DS [65,66]. Electrophysiological recordings from hippocampi of Ts65Dn mice revealed abnormal long-term potentiation and long-term depression, another indicator of hippocampal dysfunction [67,68]. In addition to hippocampal dysfunction, the Ts65Dn mouse also displays a hematopoietic phenotype featuring myeloproliferative disease comparable to that found in some DS patients. Severe thrombocytosis, hyperplasia of megakaryocytes as well as myelofibrosis associated with defective hematopoietic stem cells and myeloid progenitor cells were reported in DS [69,70,71,72]. Ts65Dn mice also exhibit cardiac defects similar to those seen in DS, suggesting that the genes responsible for this phenotype may be located within the portion of the chromosome triplicated in the Ts65Dn mouse [73]. Therefore, in spite of lacking two human chromosome 21 syntenic regions located on murine chromosome 10 and 17, the Ts65Dn mouse displays many clinically relevant features of DS and provides an excellent platform for investigating the genetic causes of DS phenotypes.
![Fig. 1. Schematic representation of the long-arm of human chromosome 21 comprised of the mouse chromosomal segments from chromosome 16, 17 and 10. The portion of chromosome 16 that is triplicated in the Ts65Dn mouse contains approximately 99 of the 170 orthologous genes [58] and harbors miR-802 sequence. According to cytogenic analysis, the loci for miR-99a, let-7c, miR-125b and miR-155 are outside the Ts65Dn triplicated segment [59]. Mrpl39 = Mitochondrial ribosomal protein L39; Zfp295 = zinc finger protein 295 (human ZNF295).](https://karger.silverchair-cdn.com/karger/content_public/journal/dne/33/5/10.1159_000330884/2/m_000330884_f01.jpeg?Expires=1716301421&Signature=ZoxX3k1u6E-k~vmijSgfyFralSiG-Dxvqyz5dqqie5gbdg15iQYXtlQqMSWGpON3OHLSHRnOD30UOh4RZrXpGnszKDjXVhtMfD9RZYjuBaUowRpfd~FNKOFc166fjMv51tK7WzM84BrQi8i0WDTGTxaDVEfndAe~tzmYomB1MTESXWfDAuGSSmn6vEOLWHONg4J3Ofcov~x1cuvzAi2k10f-vDBkCiE0dXfUSSbgwkaSAJBcuYm9YCrlJ-YIZ6D8FfW9QvpTs3Eo7MDPwnyFMSI3Gj6CScaP4Zc2dCnLf1~v5PGzDVkPgLzadpRjhRYm3dmAXKjPPtoHbrkGG62tlw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Schematic representation of the long-arm of human chromosome 21 comprised of the mouse chromosomal segments from chromosome 16, 17 and 10. The portion of chromosome 16 that is triplicated in the Ts65Dn mouse contains approximately 99 of the 170 orthologous genes [58] and harbors miR-802 sequence. According to cytogenic analysis, the loci for miR-99a, let-7c, miR-125b and miR-155 are outside the Ts65Dn triplicated segment [59]. Mrpl39 = Mitochondrial ribosomal protein L39; Zfp295 = zinc finger protein 295 (human ZNF295).
Schematic representation of the long-arm of human chromosome 21 comprised of the mouse chromosomal segments from chromosome 16, 17 and 10. The portion of chromosome 16 that is triplicated in the Ts65Dn mouse contains approximately 99 of the 170 orthologous genes [58] and harbors miR-802 sequence. According to cytogenic analysis, the loci for miR-99a, let-7c, miR-125b and miR-155 are outside the Ts65Dn triplicated segment [59]. Mrpl39 = Mitochondrial ribosomal protein L39; Zfp295 = zinc finger protein 295 (human ZNF295).
In this study, we examined the miRNA profiles in the Ts65Dn mouse model of DS and euploid control hippocampi using miRNA TaqMan arrays to elucidate the contribution of miRNAs to the DS phenotypes. We also determined the expression levels of specific miRNAs (miR-155, miR-214, miR-223 and miR-802) in the hippocampus, parietal cortex, whole blood and lung for an independent set of samples derived from Ts65Dn and euploid littermates to examine their expression levels in tissues that may be impacted by these miRNAs leading to recognized DS phenotypes. Finally, we measured the mRNA and protein expressions levels of the selected targets of miRNAs that showed significant differences in Ts65Dn as compared to euploid mice.
Methods
Ts65Dn Mice
Ts65Dn and control diploid mice were purchased from Jackson Laboratories (Bar Harbor, Me., USA). Some of the mice were obtained from in-house breeding and bred according to a previously established protocol [67]. All mice were housed in a temperature-controlled room (25°C) with free access to food and water and maintained on a 12-hour light/dark cycle. All procedures used during this study were reviewed and approved by the Uniformed Services University of the Health Sciences Institutional Animal Care and Use Committee. Measures were taken to minimize pain and discomfort and efforts were made to use the minimum number of animals while maintaining statistically valid numbers. All mice were karyotyped by Jackson Laboratories and/or by a previously described procedure [60,74]. All results reported here are from 5- to 6-week-old male mice.
RNA Isolation from Whole Blood, Lung and Brain Tissue and miRNA Assays
Mice were deeply anesthetized with isoflurane throughout all procedures. Whole blood was obtained via cardiac puncture and immediately placed into 2-ml tubes pre-filled with RNAlater supplied with the Mouse Ribopure™-Blood RNA Isolation kit (Applied Biosystems/Ambion, Austin, Tex., USA). Total RNA was isolated from whole blood using the Mouse Ribopure-Blood RNA Isolation kit according to the supplied protocol.
Immediately after the collection of whole blood, the mouse was decapitated, the brain was removed from the calvarium and the hippocampus and parietal cortex dissected out. The lungs were dissected from the thoracic cavity (as described at http://eulep.pdn.cam.ac.uk/Necropsy_of_the_Mouse/) with one lung from each mouse being placed in 10% buffered formalin for histologic review. Immediately following dissection, the hippocampi, parietal cortex and the other lung were placed in 10× volume of RNAlater (Applied Biosystems/Ambion) and stored at 4°C overnight. RNAlater was removed from the samples and the samples were then stored at –80°C until use. Total RNA was isolated from the hippocampus, parietal cortex and lung using the mirVana microRNA Isolation Kit according to the supplied protocol (Applied Biosystems/Ambion).
Reverse transcription was performed with TaqMan® microRNA Reverse Transcription Kit (Applied Biosystems, Foster City, Calif., USA) specific for each miRNA.
All quantitative real-time PCR was performed using 10 ng of input RNA per manufacturer’s recommendation except for the miR-802 assays. Due to the low expression level of miR-802, 500 ng of input RNA was used for the reported results. Gene expression was calculated relative to U6. For sequences used in the individual miRNA assays see online suppl. table 4 (for all suppl. material, see www.karger.com?doi=10.1159/000330884).
Real-Time Quantification of Mature miRNAs Using ABI TaqMan Arrays
Total RNA was isolated from the hippocampus as described above. Mature miRNA profiling was performed using TaqMan® Rodent microRNA A cards (Applied Biosystems), which contain 335 different miRNAs according to the supplied protocol without preamplification. 500 ng of input RNA was used per manufacturer’s recommendation. Briefly, multiplex reverse transcription was performed with TaqMan microRNA Reverse Transcription Kit (ABI). The RT reaction mixture contained 100 mM dNTPs (with TTP) 0.20 µl, Multiscribe Reverse Transcriptase (50 U/µl) 1.5 µl, 10× RT Buffer 0.8 µl, MgCl2 (25 mM) 0.9 µl, RNAse inhibitor (20 U/µl) 0.1 µl, 10× Megaplex RT A primers 0.8 µl and nuclease-free water to a final volume of 4.5 µl for each reaction. 500 ng of total RNA was added to each tube and incubated on ice for 5 min. Thermal cycle for cDNA was 16°C for 2 min, then 42°C for 1 min for 40 cycles, followed by heat inactivation at 85°C for 5 min in a Thermal Cycler (Applied Biosystems 2720). 100 µl of the RT reaction-specific PCR reaction mix was loaded into the ports of the TaqMan Rodent MicroRNA Panel v2.0.
Quantification of mRNA Expression
First-strand cDNA synthesis with RNA isolated from the hippocampi, whole blood and lungs, and subsequent qPCR using LUX primers (Invitrogen) was carried out on ABI 7900 using Superscript III Platinum two-step qRT-PCR kit (Invitrogen). Briefly, 250 ng of high-quality, intact RNA was used to synthesize first-strand cDNA synthesis in 20 µl reaction volume containing 2× RT Reaction Mix (10 µl) and RT enzyme mix (2 µl). The reaction mix was incubated at 25°C for 10 min and the cDNA synthesis at 42°C for 50 min. The reaction was terminated at 85°C for 5 min and then chilled on ice. Escherichia coli RNase H (2U) was added and incubated at 37°C for 20 min. qPCR was carried out in a volume of 25 µl using final concentration of 1× platinum quantitative PCR superMix-UDG, 200 nM each of LUX-labeled and unlabeled primers, 50 nM of Rox reference dye and 2.5 µl of cDNA from the first-strand synthesis reaction as mentioned above. Cycling reaction program was followed as: 50°C for 2 min (UDG incubation), 95°C for 2 min, and 40 cycles of 95°C for 15 s and 60°C for 1 min. The real-time PCRs were performed in triplicate for target genes (Mecp2, Ship1, Ezh2 and Creb1) and normalized to β-actin expression. Pri- and pre-miR-155 expression was analyzed by SYBR® green-based real-time PCR according to the manufacturer’s protocol (Invitrogen). The primers used for the pri-miR-155 assay were forward: 5′-GAC ACA AGG CCT GTT ACT AGC AC-3′, reverse: 5′-GTC TGA CAT CTA CGT TCA TCC AGC-3′ and those used for pre-miR-155 assay were forward: 5′-GCT AAT TGT GAT AGG GGT TTT GG-3′, reverse: 5′-GTT AAT GCT AAC AGG TAG GAG TC-3′ (for primer sequence used in these experiments, see online suppl. table 4).
Western Blot Analysis
The hippocampal tissues from Ts65Dn and euploid mice were homogenized in RIPA buffer. The homogenates were then centrifuged and the supernatants collected. Total protein concentration was determined using BCA™ protein assay (Thermo Scientific, Hudson, N.H., USA) and Fluostar Optima microplate spectrophotometer from BMG Labtech (Offenburg, Germany). MeCP2 and EZH2 levels were determined using Western blot analysis. Briefly, protein samples were separated by electrophoresis on 4–12% Nupage gels (Invitrogen) following the manufacturer’s instructions. The proteins were transferred from the Nupage gels to polyvinylidene difluoride membranes (Pall Corporation, Ann Arbor, Mich., USA) and hybridized with either anti-MeCP2 (catalog No. 07-013; Millipore), anti-EZH2 (catalog No. 5246; Cell Signaling) or anti-GAPDH (catalog No. AB8245; Abcam) primary antibodies. After appropriate washing procedures, the membranes were incubated with goat anti-rabbit secondary antibody (catalog No. 170-6515; BioRad) or anti-mouse secondary antibody (catalog No. 172-1011; BioRad). The Western blot signals were detected using Fujifilm LAS-3000 Imager (Fujifilm, Stamford, Conn., USA).
Statistical Analysis
Statistical analysis for cycle threshold (Ct) values generated in TaqMan array experiments was performed using Real Time Statminer software (Intergromics, Philadelphia, Pa., USA) and HTqPCR package from the bioconductor project for open source data analysis tools (http://www.bioconductor.org/help/bioc-views/release/bioc/html/HTqPCR.html). Normalization of Ct values was performed using the most stable reference (housekeeping) genes selected from a set of tested candidate reference genes using the Genorm algorithm, which computes an endogenous control based on geometric averaging of multiple reference genes [75]. Based on the Genorm analysis, snoRNA-202 and snoRNA-135 were found to be most stable genes in the dataset. Ct values were filtered to allow removal of genes which had undetermined expression (Ct >36) in 5 or more samples from each group. After applying these filter and quality checks, 184 genes were considered for further analysis. Differences in the expression level of miRNA between Ts65Dn and euploid control mice were computed using the parametric test (Limma), followed by Benjamini-Hochberg false discovery rate p value adjustment. The adjusted p value cutoff was set at 0.05. Statistical analysis of individual miRNA assays was performed using GraphPad Prism software v4.0a. A one-tailed unpaired t test was performed on the data and statistical significance cutoff of p < 0.05 was considered.
Bioinformatics Analysis
Target prediction for the best findings from miRNA expression analysis was performed using bioinformatics approaches. The following criteria were considered for target prediction: (1) sequence match; (2) presence of multiple binding sites for miRNA in the gene; (3) conservation of miRNA sequence across multiple species; (4) accessibility of miRNA binding sites based on the mRNA secondary structure, and (5) localization of sites in the untranslated region of the gene. An in-house PERL program was developed to identify the potential targets. The program utilizes publicly available data from the following five resources: (1) microrna.org (http://www.microrna.org/), (2) mirDB (http://mirdb.org/miRDB/), (3) RNA22 (http://cbcsrv.watson.ibm.com/), (4) TargetScan (http://www.targetscan.org/), and (5) microT (http://diana.cslab.ece.ntua.gr/microT/) and incorporates the above-mentioned criteria. We selected gene targets for miRNA only when they satisfied all five criteria mentioned above. A conservative search strategy was employed to reduce signal-to-noise ratio by selecting an miTG score cutoff of 13 based on predictions by the microT server. Functional analysis of gene targets was performed using Ingenuity analysis software (Ingenuity Inc., Redwood City, Calif., USA).
Lung Histopathology
Light-microscopic examination was performed on all lung tissues. A representative sample of the lung was fixed in 10% neutral buffered formalin, embedded in paraffin wax and sectioned at approximately 5 µm. Sections were routinely stained with hematoxylin and eosin and examined by light microscopy. In addition, special histochemical staining was performed using Masson’s trichrome. Masson’s trichrome is used to highlight and contrast collagenous connective tissue from other mesenchymal tissues such as smooth and skeletal muscle. In doing so, hyperplastic lesions within the peribronchiolar myocytes and the smooth muscle of the arterioles and connective tissue deposition within the alveolar interstitium would have been accentuated. The histochemical special stains were performed by routine methods [76].
Results
Ts65Dn Segment of Murine Chromosome 16
As stated previously, not all of the genes found on human chromosome 21 are triplicated in the Ts65Dn model of DS (fig. 1). Bioinformatic and cytogenetic analysis has indicated that miR-802, found on human chromosome 21, is present on the triplicated segment Ts65Dn of chromosome 16 (fig. 1) [58,59,77]. However, the miR-155 host gene remains outside of the triplicated Ts65Dn murine segment (see Jackson Laboratories web site http://www.informatics.jax.org/searches/accession_report.cgi?id=MGI:104219).
Differential Expression Analysis of Ts65Dn miRNAs Using TaqMan Arrays
miRNA profiling using TaqMan arrays was performed to identify miRNA expression variation in the Ts65Dn hippocampus (n = 8 euploid, n = 7 trisomy) as compared with euploid. Eleven miRNAs were found to be significantly differentially expressed between Ts65Dn and euploid samples (p < 0.05), with 6 overexpressed and 5 underexpressed in the trisomy group (fig. 2). Only miR-210 and miR-7b showed significantly differential expression levels after correction for false discovery rate. Other prominent findings include overexpression of miR-155, which is located on mouse chromosome 16 and has previously been shown to be overexpressed in DS as well Ts65Dn hippocampus and cortex [31,32]. miR-802 was filtered out from the analysis during quality check because of its low expression in the hippocampus (see Methods).
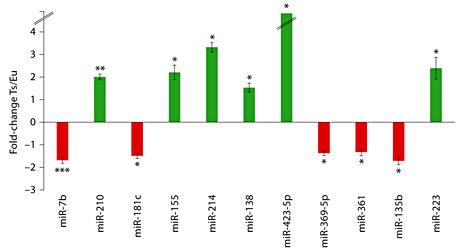
miRNAs differentially expressed in Ts65Dn hippocampus with fold-change for the 11 best findings in terms of statistical significance. Upregulation of miRNAs in Ts65Dn is represented by green bars and downregulation by red bars. Broken lines in bar for miR-423-5p indicate that this miRNA was detected in Ts65Dn but not in the control euploid hippocampus. The error bars represent the mean ± SEM utilizing 8 euploid and 7 trisomy mice samples. * p < 0.05, ** p < 0.01, *** p < 0.005.
miRNAs differentially expressed in Ts65Dn hippocampus with fold-change for the 11 best findings in terms of statistical significance. Upregulation of miRNAs in Ts65Dn is represented by green bars and downregulation by red bars. Broken lines in bar for miR-423-5p indicate that this miRNA was detected in Ts65Dn but not in the control euploid hippocampus. The error bars represent the mean ± SEM utilizing 8 euploid and 7 trisomy mice samples. * p < 0.05, ** p < 0.01, *** p < 0.005.
A hierarchical clustering heatmap demonstrated that expression profiles of 11 miRNAs determined by TaqMan arrays can separate samples into euploid and trisomy groups (fig. 3). A supervised approach was used for clustering with p value cutoff set at 0.05 and Pearson’s correlation distance measures. From the array results, we searched for miRNAs that had 2-fold differences in expression levels between the two groups (online suppl. table 1). We found that 5 out of 6 miRNAs overexpressed in Ts65Dn mouse satisfy these criteria, including miR-155, miR-214 and miR-223. However, none of the miRNAs significantly downregulated in trisomy had greater than 2-fold differences in expression levels.

Heatmap of differentially expressed miRNAs in Ts65Dn hippocampus. Heatmap was generated by supervised hierarchical clustering of normalized Ct values of miRNAs with altered expression in Ts65Dn hippocampus compared with normal euploid. A p value cutoff of 0.05 was set to filter miRNAs used for clustering and Pearson’s correlation was used as the distance metric for the underlying computations. Sample names are presented in columns and miRNAs in rows. A color gradient represents the relative miRNA expression levels as indicated in the legend box. It is demonstrated that expression levels of these miRNAs can separate samples into biologically interpretable groups.
Heatmap of differentially expressed miRNAs in Ts65Dn hippocampus. Heatmap was generated by supervised hierarchical clustering of normalized Ct values of miRNAs with altered expression in Ts65Dn hippocampus compared with normal euploid. A p value cutoff of 0.05 was set to filter miRNAs used for clustering and Pearson’s correlation was used as the distance metric for the underlying computations. Sample names are presented in columns and miRNAs in rows. A color gradient represents the relative miRNA expression levels as indicated in the legend box. It is demonstrated that expression levels of these miRNAs can separate samples into biologically interpretable groups.
miRNAs Expression in Ts65Dn Hippocampus, Parietal Cortex and Whole Blood
In order to validate our array results and confirm findings of the previous studies, we performed individual assays for four miRNAs: miR-155, miR-802, miR-214 and miR-223. An independent set of samples (n = 5 for each hippocampus and blood, n = 4 for parietal cortex) was selected, and individual assays were performed on RNA extracted from Ts65Dn hippocampus, parietal cortex, and whole blood.
Our results confirmed previous findings [32] and revealed overexpression of miR-155 (p = 0.011, 4.5-fold increase) and miR-802 (p = 0.001, 3.5-fold increase) in the hippocampus of Ts65Dn mice (fig. 4a). We also found a 4-fold increase for miR-155 (p = 0.04) and a 2.5-fold increase for miR-802 (p = 0.05) in the blood of trisomy mice compared to the control group. miR-214 was also found 2.5-fold overexpressed (p = 0.01) in hippocampus of the trisomy group, but no significant expression difference was observed in the blood.

Comparison of miRNA and mRNA expression in Ts65Dn mice as compared to the control euploid. Individual miRNA assays for three miRNAs were performed with RNA extracted from hippocampus (5 Ts65Dn and 5 euploid) (a) and whole blood (5 Ts65Dn and 5 euploid) (b). (c) Pri- and pre-miR-155 was quantified by real-time qPCR on RNA samples derived from hippocampus (5 Ts65Dn and 5 euploid). (d) mRNA was quantified by real-time qPCR on RNA samples derived from hippocampus (5 Ts65Dn and 5 euploid). (e) Ship1 mRNA was quantified by real-time qPCR on RNA samples derived from whole blood and lungs (5 Ts65Dn and 5 euploid). The error bars represent the mean ± SEM of three independent experiments utilizing 5 mouse hippocampal samples. * p < 0.05, ** p < 0.01, *** p < 0.005.
Comparison of miRNA and mRNA expression in Ts65Dn mice as compared to the control euploid. Individual miRNA assays for three miRNAs were performed with RNA extracted from hippocampus (5 Ts65Dn and 5 euploid) (a) and whole blood (5 Ts65Dn and 5 euploid) (b). (c) Pri- and pre-miR-155 was quantified by real-time qPCR on RNA samples derived from hippocampus (5 Ts65Dn and 5 euploid). (d) mRNA was quantified by real-time qPCR on RNA samples derived from hippocampus (5 Ts65Dn and 5 euploid). (e) Ship1 mRNA was quantified by real-time qPCR on RNA samples derived from whole blood and lungs (5 Ts65Dn and 5 euploid). The error bars represent the mean ± SEM of three independent experiments utilizing 5 mouse hippocampal samples. * p < 0.05, ** p < 0.01, *** p < 0.005.
It has previously been shown that miR-155 and miR-802 are overexpressed in prefrontal cortex [32], but their levels in the parietal cortex have not been previously reported. Our results did not show statistically significant changes in miR-155 (2-fold increase, p = 0.25) and miR-802 (1.9-fold increase, p = 0.15) in the parietal cortex of the Ts65Dn mouse. It appears that small sample size and sample variability might affect the reported outcome more than in hippocampus and in whole blood.
It is well known that DS subjects are at increased risk for the development of various leukemias as well as transient myeloproliferative disease with miR-155 and miR-223 likely contributing to the development of leukemia and altered hematopoiesis [78,79,80,81]. In support of these observations, we found that miR-155, miR-223, miR-214 and miR-802 were significantly overexpressed in blood of Ts65Dn mice when compared to euploid controls (fig. 4b).
Results of Primary and Precursor miR-155 Analysis
Mature miRNAs are produced from long primary transcripts through sequential cleavages by the Microprocessor and Dicer complexes. The function and expression of mature miRNAs is controlled by mechanisms that regulate the processing of primary (pri-) and precursor (pre-) miRNAs. Increased expression of miR-155 may contribute to hippocampal deficit in Ts65Dn; therefore, we decided to examine the biogenesis leading to its upregulation. We analyzed the expression of miR-155 precursors: pri- and pre-miR-155 in Ts65Dn hippocampus. As depicted in figure 4c, there is a ∼3-fold upregulation of pri-miR-155 as well as pre-miR-155 (p < 0.05). Since we find ∼4-fold upregulation of mature miR-155 (fig. 4a, b), the data strongly suggest that the enhanced expression of miR-155 is due both to increased synthesis of its precursors as well as enhanced processing.
mRNA and Western Blot Analysis of Selected Putative Targets of Overexpressed miRNAs
In order to verify the functional outcome of miRNA overexpression, we selected three genes with important implications for hippocampal synaptic plasticity. These include potential targets of miR-214 (Ezh2, enhancer of Zeste homolog 2) and miR-155 (Mecp2 and Ship1). We also selected Creb1, which is a downstream target of the transcriptional factor Mecp2, and was previously reported to be decreased in Ts65Dn hippocampus [32]. We found significant attenuation of mRNAs for Ezh2, Mecp2 and Ship1 in Ts65Dn hippocampus (fig. 4d). Western blot results confirmed attenuation of MeCP2 and EZH2 protein expression in the hippocampus as well (online suppl. fig. S1). Interestingly, in correlation with miR-155 expression in the Ts65Dn, Ship1 expression was decreased in whole blood but was not affected in the lungs (fig. 4e).
Functional Analysis of Target Genes
A total of 12 miRNA genes were used for the target gene search, which includes 11 significant hits from array analysis and miR-802. miR-802 was added to the list because it was found to be overexpressed in the hippocampus and blood in our study using individual assays. In addition, there is prior evidence for its overexpression in Ts65Dn hippocampus [32] and it is located on the triplicated region of the mouse chromosome 16 [31]. Gene targets were predicted using a combination of in-house and publically available computational tools (see Methods). A strict search criterion was employed to eliminate false-positive findings, which involved selection of targets only when they were independently predicted by all five resources used, namely microRNA.org [82], RNA22 [83], microT [84], miRDB [85] and TargetScan [86]. We found 1,442 potential binding sites for the 12 miRNAs, which map to 310 mouse genes (online suppl. table 2). Functional analysis was performed using Ingenuity pathway analysis software, which indicates that our gene list is significantly enriched in the neurological disorder category. A number of genes were mapped to signaling pathways, including the MAP kinases (MAPK1) and BDNF receptor (NTRK2). The physiological functions potentially impacted due to association with the target genes (p < 0.005) include nervous system development, behavior and tissue development (fig. 5). Enrichment of biological processes in terms of Gene Ontology categories with respect to the target genes was determined using the DAVID Bioinformatics Resources. Again, many developmental and differentiation-related cellular processes appeared as the best hits (p < 0.005), most of which were associated with the central nervous system (fig. 6). Interesting targets (fig. 7) that have been previously implicated in Ts65Dn hippocampal deficit include Mecp2 [32], protein kinase C [87], GSk3b [88] and adenylate cyclase [87].
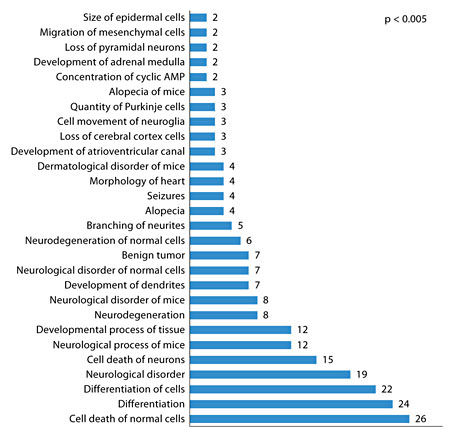
Enrichment of function categories (molecular and physiological function as well as involvement in diseases) in the target genes was determined using Ingenuity IPA software (p < 0.005). A number of functions linked to DS phenotype were observed.
Enrichment of function categories (molecular and physiological function as well as involvement in diseases) in the target genes was determined using Ingenuity IPA software (p < 0.005). A number of functions linked to DS phenotype were observed.
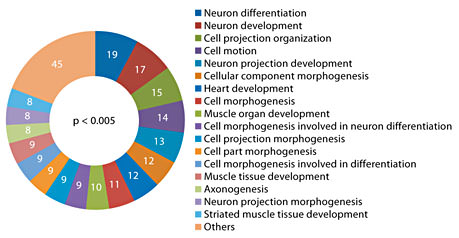
Enrichment of biological processes in terms of Gene Ontology (GO) categories with respect to the target genes (p < 0.005) for differentially expressed miRNAs. GO categories for each target gene were determined using the DAVID Bioinformatics Resources (http://david.abcc.ncifcrf.gov/). The number of genes in each GO category is indicated within the plot. Nervous system- and development-related biological process had maximum representation among the target genes.
Enrichment of biological processes in terms of Gene Ontology (GO) categories with respect to the target genes (p < 0.005) for differentially expressed miRNAs. GO categories for each target gene were determined using the DAVID Bioinformatics Resources (http://david.abcc.ncifcrf.gov/). The number of genes in each GO category is indicated within the plot. Nervous system- and development-related biological process had maximum representation among the target genes.
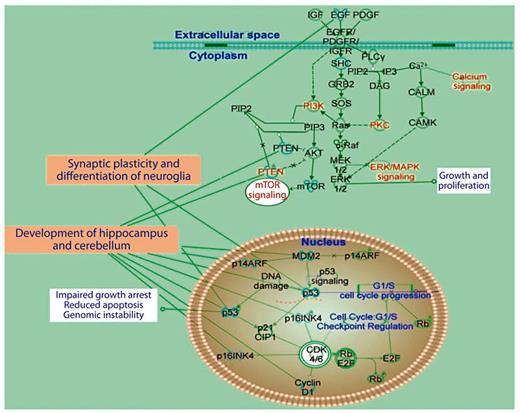
Schematic representation of signaling pathways involved in synaptic plasticity and neuroglia functions with associated molecules and cellular tasks (analysis based on Ingenuity IPA software). A number of component genes in the pathway are potential contributors to the DS phenotype.
Schematic representation of signaling pathways involved in synaptic plasticity and neuroglia functions with associated molecules and cellular tasks (analysis based on Ingenuity IPA software). A number of component genes in the pathway are potential contributors to the DS phenotype.
miRNA Expression and Histopathology of Ts65Dn Lungs
Increased incidence of persistent pulmonary hypertension of the newborn with DS suggests that histopathological changes may be present in Ts65Dn lungs [89,90]. Histopathological analysis of lung samples from both euploid and Ts65Dn mouse littermates were evaluated in a blind manner. The pulmonary interstitium was assessed for increased deposition of collagen, suggestive of pulmonary fibrosis, and the pulmonary vasculature was examined for changes suggestive of pulmonary hypertension, such as smooth muscle hypertrophy, endothelial proliferation and collagen accumulation. Comparative histopathological evaluation of all submitted tissues did not reveal any significant differences between the euploid and Ts65Dn mice (online suppl. fig. S2). The average vessel wall width of medium-sized pulmonary vessels was approximately 41.5 µm in Ts65Dn and 42 µm in euploid mice (online suppl. fig. S2). Furthermore, there was no evidence of increased deposition or abnormal accumulation of collagen in any of the submitted samples (online suppl. fig. S2). Expression levels of miR-155, miR-802, miR-204, miR-214 and miR-223 were similar in Ts65Dn and euploid lungs (online suppl. table 3, n = 5 for each). Ship1 (target of miR-155) expression was normal (fig. 4e) in Ts65Dn lung tissue, in contrast to what we found in the hippocampus and the whole blood but correlated with normal expression of miR-155.
Discussion
The DS phenotype is complex, variable and results from the altered expression of multiple genes on chromosome 21. As of yet, no single gene or region of chromosome 21 has been identified that can explain all of the features of the DS phenotype [7,19]. More recently there has been an increased interest in the area of epigenetics, specifically in miRNA expression and its potential role in health as well as disease states. Interestingly, it has been reported that five miRNAs located on chromosome 21 were overexpressed in DS brain and heart tissue [30,31,32], potentially providing a molecular basis for the complex features underlying DS phenotypes. The gene encoding miR-802 is present in the triplicated Ts65Dn chromosomal segment (fig. 1); however, we found significant dysregulation of multiple miRNAs, for which host genes are located outside the triplicated segment including miR-155. As the majority of our findings are specific for the hippocampus, we will first focus on their implications for hippocampal deficits reported in DS subjects and Ts65Dn mice. We will then discuss the potential impact of dysregulation of whole blood miRNAs on hematological defects and tumorigenesis recognized as part of the DS phenotype [65,91,92,93,94].
DS-Related Phenotypes and Differentially Expressed Ts65Dn Hippocampal miRNAs
Using ABI TaqMan arrays, we found differential expression of 11 miRNAs between Ts65Dn and euploid hippocampus. Among these, miR-210, miR-155, miR-214, miR-138, miR-423-5p and miR-223 showed a significant increase in the Ts65Dn hippocampus, whereas reduced levels of miR-7b, miR-181c, miR-369-5p, miR-361 and miR-135b were observed as compared to euploid control hippocampus. Furthermore, expression evaluation of miR-155, miR-223, miR-214 and miR-802 using individual assays exhibited higher levels of all these miRNAs in the Ts65Dn hippocampus, thereby confirming previous reports [32] as well as our results derived from ABI TaqMan arrays. These miRNAs target various genes located within and outside the triplicated Ts65Dn chromosomal segment. These genes may contribute to the hippocampal deficits, thus affecting cognitive abilities of DS subjects by altering the gene expression profile and transcriptional networks underlying synaptic plasticity [95,96,97].
miR-155 has emerged as a critical regulator of not only immune cell development but also exhibits potential to impact neurophysiological function and contribute to DS neurological symptoms. miR-155 acts via targeting Src homology-2 domain-containing inositol 5-phosphatase 1 (SHIP1), an inhibitor of the PI3K/Akt signaling pathway [98]. Our results confirm downregulation of Ship1 mRNA, which would be consistent with hyperactivation of the PI3K/Akt signaling pathway in Ts65Dn hippocampus. In fact, we previously reported an increase in phosphorylated Akt level in Ts65Dn hippocampus and the lack of an increase in Akt phosphorylation that normally follows high-frequency stimulation of hippocampal pathway [87]. We concluded that these changes could lead to abnormal long-term synaptic plasticity, long-term potentiation and long-term depression, and the result presented here could offer a new rational framework for our previous findings.
We present here evidence that upregulation of mature miR-155 in Ts65Dn hippocampus appears to be caused by abnormal expression of precursors as well as enhanced processing of miR-155. These results suggest abnormal miR-155 host gene/BIC biogenesis, similar to previous reports in chronic lymphocytic leukemia [99] and cystic fibrosis [Bhattacharyya and Biswas, pers. commun.]. This dysregulation can be triggered by protein kinase C and nuclear factor ĸB, or other proinflammatory factors [100]. In parallel to the hippocampal level, miR-155 was also upregulated in the Ts65Dn blood. The correlation of selected miRNAs in hippocampus and blood is intriguing and it highlights their role as potential biomarkers as previously reported in rat models of brain injury [101].
A recent report shows that activation of miR-214 transcription reduces protein expression of Pcr2 (catalytic subunit of histone methyltransferase of polycomb repressive complex 2) and Ezh2 [102]. Attenuation of Ezh2 activity may impact the self-renewal and multipotency of cortical/hippocampal progenitor cells and alter their neurogenic potential [103,104]. In fact, we previously reported developmental alterations in Ts65Dn cortex and hippocampus that are consistent with attenuation of Ezh2 function in Ts65Dn mice [60,61]. In addition, abnormal adult neurogenesis has also been found in the Ts65Dn brain [105,106]. miR-214 has been reported to regulate su(fu) mRNA, encoding a negative regulator of sonic Hedgehog signaling [107]. This, combined with a previously reported defective cerebellar response to mitogenic Hedgehog signaling in Ts65Dn mice [108] (thus attributed to the cerebellar phenotype in DS [109]), supports their potential contribution to abnormal adult hippocampal neurogenesis in Ts65Dn [105,110] and suggests aberrant neuronal differentiation [111,112]. Interestingly, overexpression of miR-223 in the hippocampus was found days after traumatic brain injury (TBI) [113] and in neutrophils after spinal cord injury [114]. Significant upregulation of miR-155 after TBI was also reported [113], suggesting that together with miR-223, they may play a role in the onset of abnormal hippocampal synaptic plasticity following TBI [115] and potentially contribute to similar deficits reported in Ts65Dn mice [92].
Another upregulated miRNA, miR-138, may be significant for hippocampal activity as a functional screen has implicated miR-138-dependent regulation of the depalmitoylation enzyme APT1 in dendritic spine morphogenesis, a phenomenon associated with long-lasting forms of memory [116].
Among underexpressed miRNAs in Ts65Dn hippocampus, miR-7b, has been shown to downregulate the expression of the Fos [117] gene, which is a marker for neuronal activity, and is known to regulate a wide range of physiological processes in the brain affecting functions such as cell signaling, proliferation and synaptic plasticity. Increased levels of Fos mRNA were found in frontal, parietal and temporal cortex of DS subjects [118]. Decreased Fos expression in the individual brain regions may be linked to increased apoptosis, neurodegeneration, or abnormal network activity contributing to DS pathology.
We have found a dramatic increase in the expression of miR-423-5p in the Ts65Dn hippocampus; however, there is no published evidence on its role in this area of the brain. Similarly, there is no published evidence for the hippocampal role of downregulated miR-369-5p or miR-361.
Abnormal Expression of miRNAs in Ts65Dn Blood
A well-established feature of DS is the increased risk for the development of leukemia and abnormal hematopoiesis [119]. It has been shown that miRNA play important roles in normal hematopoiesis as well as oncogenesis [80,81,120]. Specifically, abnormal expressions of miR-155 and miR-223 have been implicated in the development of multiple leukemias [78,79,121,122,123]. While the role of miRNA in leukemia in DS subjects has been explored [119,124], no one has determined the expression level of miRNA in blood from the Ts65Dn mouse. Our results show elevated expression of miR-155, miR-223, miR-214 and miR-802 in Ts65Dn blood in comparison to euploid control. While the exact targets of miR-155, miR-214, miR-223 and miR-802 in the hematopoietic system remain to be determined, their overexpression suggests a possible role for miRNA in the determination of DS hematopoietic features and their interaction with other abnormally expressed miRNAs may contribute to the variability of this DS phenotype.
Specific knockdown of SHIP1 in the hematopoietic system of mice following retroviral delivery of a miR-155-formatted SHIP1 siRNA cassette has been shown to result in a myeloproliferative disorder [98] reported in DS [125]. In addition to its role in the hippocampus, miR-223, which we found upregulated in Ts65Dn blood, has been implicated in myeloid differentiation and its overexpression has been associated with a subset of adult T cell acute lymphoblastic leukemia [126], a highly prevalent trait in DS subjects [125]. miR-223 is also the highest expressed megakaryocytic miRNA in primary myelofibrosis and essential thrombocythemia [127]. miR-223 can trigger normal granulopoiesis and is an important element of the autoregulatory negative feedback loop in acute myeloid leukemia. Suppression of miR-223 has been found in cells from acute myeloid leukemia patients [79,123], suggesting that overexpression of this miRNA may have a significant contribution to hemopoietic DS deficits.
Tumorigenesis
Several studies have reported miR-210 as one of the highly upregulated miRNAs in hypoxic cells and have demonstrated its importance for cell survival [128]. Also, HIF-1α-dependent miR-210 regulates the expression of HOXA1, HOXA9 and FGFRL1 genes, and acts as a rheostat for cellular adaptation and survival by inhibiting tumor initiation [129]. These findings combined with the increased miR-210 levels observed in Ts65Dn hippocampus in the present study would be consistent with the decreased incidence of solid cancers observed in DS subjects [125,130].
Normal miRNAs in Ts65Dn Lungs
In addition to aberrancies in hematopoiesis, it has recently been determined that infants with DS are at an increased risk for the development of persistent pulmonary hypertension of the newborn [8,9]. Currently, there is little data to suggest why DS infants would be at an increased risk for developing this disease process. A recently published study has shown that downregulation of miR-204 in pulmonary artery smooth muscle cells is correlated with pulmonary hypertension [131]. Our study, however, showed that miR-155, miR-802, miR-223, miR-214 and miR-204 were normal in Ts65Dn lungs and histopathological changes were absent in Ts65Dn lungs.
Downstream Impact of Abnormal miRNAs on Nonchromosome 21 Genes
Implementing bioinformatic analysis, we attempted to determine the potential target genes of differentially regulated miRNA genes identified in Ts65Dn. The up- or downregulated hippocampal miRNAs were found to impact more than 300 genes in the hippocampus. Interestingly, most of them were not located on the triplicated Ts65Dn segment or their respective human chromosome 21 orthologs. Analyses of the global transcriptome of murine models of DS had revealed an average 1.5-fold overexpression of genes presented on the extra chromosomal segments in cortex, cerebellum and other organs [13,14,132,133]. However, a significant number of transcripts with only two gene copies were significantly altered as well (e.g. in Ts1Cje cerebellum this number varied between 3 and 5% depending on developmental stage). The reason for this variation has been ascribed to complex interactions between gene networks and the presence of transcriptional factors on the duplicated segment. Our findings presented here and recent reports on abnormal expression of miRNAs in Ts65Dn and human DS tissues [30,31,32] strongly suggest that some of the changes to the transcriptome coded by nonchromosome 21 genes can be ascribed to interactions between chromosome 21 miRNAs and their transcriptional targets on other chromosomes.
As far as we know global transcriptional analysis has not yet been reported in Ts65Dn hippocampus. Therefore, the results presented here cannot be directly verified with the exception of the few specific targets selected by us. We have examined the mRNA expression of three target genes including Ship1, Mecp2, Creb1 (indirect target) and Ezh2. Significantly reduced levels of mRNA for Ship1, Mecp2, Creb1 and Ezh2 were observed in Ts65Dn hippocampus, and further corroborated with decreased protein levels.
Results of pathway analysis depicted in figure 6 revealed that signaling pathways underlying synaptic plasticity are likely affected by differentially expressed hippocampal miRNAs. The important elements include pathways downstream of platelet-derived growth factor receptors, epidermal growth factor receptors and insulin-like growth factor receptors that are involved in regulation of cell growth, proliferation, neuroglia differentiation, blood vessel formation, apoptosis, cancer, genomic instability and neuronal development (neurogenesis, synaptogenesis and myelination). The results of this bioinformatic analysis (fig. 7) also suggest dysregulation of intracellular pathways such as PI3K, protein kinase C and ERK/MAPK, which we found abnormal in our previous study related to signaling pathways and long-term synaptic plasticity in Ts65Dn hippocampus [87]. PTEN (phosphatase and tensin homolog) and m-TOR (mammalian target of rapamycin) are involved in regulation of PI3K activity and redox status that is abnormal in Ts65Dn hippocampus [87,134] and DS [135,136]. Nuclear pathways that can further impact Ts65Dn phenotype include cyclin-dependent kinase inhibitor p21, E2F transcription factor that play a major role in G1/S transition [137,138] and tumor suppression hub protein p53, which has been implicated in the inhibition of angiogenesis as shown in animal models of DS [93,139]. Some of the intracellular and nuclear targets also contribute to known cerebellar deficits observed in DS and Ts65Dn [108,140,141].
In summary, we have presented a set of important results that further implicates abnormal miRNA expression as a potential cause of DS hippocampal and nonhippocampal deficits. Additional neurodevelopmental studies are warranted to better elucidate miRNA roles in abnormal neurogenesis and the abnormal balance between inhibitory and excitatory networks observed in the Ts65Dn brain [61,142]. In addition, profiling of miRNA expression will further define their contribution to known DS traits and potentially lead to novel treatment options for the diseases that affect DS subjects.
Acknowledgements
J.K.W. would like to thank Dr. Merrily Poth, Dr. Russell Moores and Dr. Jay Kerecman for their support and encouragement during her fellowship at Dr. Galdzicki’s lab. We would like to thank Dr. Nathan Cramer for his critical suggestions during the course of the work and his excellent comments on the manuscript. We also would like to thank Dr. Harvey Pollard for his stimulating discussion.
This research was funded in part by NIH R01 HD05780 (Z.G.), Lejeune Foundation (Z.G.) and intramural grants from the Uniformed Services University of the Health Sciences (J.K.-W. and Z.G.).
References
J. Keck-Wherley and D. Grover contributed equally to this paper.
