

Abstract
Although neuroendocrine tumors (NET) are a relatively rare malignancy, the reported incidence is increasing, and some of the current treatment options are limited in their efficacy. Standard first-line therapy for metastatic small bowel NET includes somatostatin analogs. Although these agents can provide symptom relief and can delay disease progression in many patients, ultimately, new treatments are required for patients with progressive disease. In recent years, there has been considerable interest in developing agents specifically targeted against some of the pathways known to be involved in cancer cell growth, survival and invasion. In 2011, the mammalian target of rapamycin (mTOR) inhibitor everolimus and the tyrosine kinase inhibitor sunitinib were approved for the treatment of pancreatic NET. Clinical trials evaluating novel targeted agents are ongoing, both as single agents and in combination regimens. We review the current clinical status of these potential new treatments and highlight those with particular promise for the management of well-differentiated NET.
Introduction
Neuroendocrine tumors (NET) are relatively rare malignancies that are observed most commonly in the gastrointestinal tract and the bronchopulmonary system [1]. Gastroenteropancreatic (GEP)-NET can arise anywhere within the gastrointestinal tract but are most frequently seen in the small bowel, rectum and pancreas [2]. The etiology of GEP-NET remains elusive, but the reported incidence appears to be increasing [2]. The first-line medical therapy generally consists of a somatostatin analog (SSA) such as octreotide, which can control symptoms related to hypersecretion, can inhibit tumor growth and can improve progression-free survival (PFS) [3,4].
Recent studies have identified new chemotherapy combinations, such as capecitabine and temozolomide, which appear to have high efficacy in treating patients with pancreatic NET (pNET) [5]. Early trials had indicated that streptozocin in combination with either 5-fluorouracil or doxorubicin was effective in the treatment of pNET, but subsequent studies suggested that the benefits observed with the use of streptozocin-containing chemotherapy were likely overestimated. In addition, there has been considerable interest and effort in conducting trials using novel targeted agents. In 2011, two phase III trials, one evaluating everolimus and the other evaluating sunitinib, reported significant improvements in PFS in patients with advanced pNET [6,7]. In this article, we focus on the current status of targeted drug therapy in the treatment of patients with GEP-NET, with special emphasis on the various biological pathways involved.
The mTOR Pathway
The mammalian target of rapamycin (mTOR) is an intracellular serine-threonine kinase that serves as an integral part of several signaling pathways, including vascular endothelial growth factor (VEGF), insulin-like growth factor receptor (IGFR) and phosphoinositol 3-kinase (PI3K)-Akt [8]. mTOR has a role in the regulation of cell growth and proliferation, cell metabolism, protein synthesis and apoptosis and is upregulated in various cancers as a result of excessive stimulation by cytokines and growth factors located upstream (fig. 1) [8]. Two mTOR inhibitors, temsirolimus and everolimus, both rapamycin derivatives, have been evaluated in clinical trials for the treatment of patients with multiple types of malignancies including NET. No reliable biomarkers are available for predicting responses to therapy with mTOR inhibitors. Such markers would be of value to the treating clinician. Several potential markers have emerged, including chromogranin A, neuron-specific enolase and cyclin D, that may sensitize the tumor cells to mTOR therapy. Conversely, Bcl2 overexpression and mutations of KRAS may predict resistance. These markers must be validated in future clinical trials [9,10].
![Fig. 1. The mTOR signaling network. Arrows represent activation, bars represent inhibition. mTOR signaling regulates multiple critical cellular processes by integrating energy and nutrient status and PI3K/Akt signaling induced by growth factors and insulin. Adapted from Meric-Bernstam et al. [8].](https://karger.silverchair-cdn.com/karger/content_public/journal/ocl/83/3/10.1159_000339539/2/m_000339539_f01.jpeg?Expires=1716365240&Signature=lfqupRLKxekxGBBMB9XmI0rk-WmllwN4BiINXCEDWNXR4YJCHwnfh2WT~zNwoOZf9cN~8dERR8xBN6xGozDPNTAo4NZjWkGR-IfQSchOY-EjYi1n1t5hLadAwXUbIfhmmiA9dQ6069cCN6~RPJx~WDZWbkmVK01NjqLoaUt2S0vBQOon6MTViXjkcGi98pH7yMCol~Il3XuTnw6yNOdt8I4enTF9UMETvWNMKGLCO5DGcf69rR6wqK7jH3JB~fznweQT~qeW9mT5O2EpzwLJmF7dqP~MzoZboSWJIKnYmFx5f8ZDBIQ5R1n0S2pXpbFAmzbuIEb-wlY0V3Z5giwUkQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
The mTOR signaling network. Arrows represent activation, bars represent inhibition. mTOR signaling regulates multiple critical cellular processes by integrating energy and nutrient status and PI3K/Akt signaling induced by growth factors and insulin. Adapted from Meric-Bernstam et al. [8].
The mTOR signaling network. Arrows represent activation, bars represent inhibition. mTOR signaling regulates multiple critical cellular processes by integrating energy and nutrient status and PI3K/Akt signaling induced by growth factors and insulin. Adapted from Meric-Bernstam et al. [8].
Temsirolimus
Temsirolimus is approved by the US Food and Drug Administration (FDA) for the treatment of patients with advanced renal cell carcinoma, and it has been evaluated in a phase II study of 37 patients with advanced NET [11]. However, because of the limited objective response rate of 5.6%, further monotherapy studies with this agent were not pursued [11]. A phase II study of temsirolimus plus the antiangiogenic monoclonal antibody bevacizumab is underway in patients with advanced NET or related malignancies (NCT01010126; www.clinicaltrials.gov). Patient accrual for this study is nearing completion, but it will be some time before the outcomes are known.
Everolimus
Everolimus was initially evaluated in combination with octreotide long-acting repeatable (LAR) in a phase II study enrolling 60 patients with low- to intermediate-grade NET [12]. A response rate of 20% was demonstrated, together with a median PFS of 60 weeks and a 1-year overall survival rate of 83% [12]. A subsequent phase II, nonrandomized, open-label clinical trial [RAD001 in Advanced Neuroendocrine Tumors (RADIANT-1)] [13] evaluated the use of everolimus in 160 patients with metastatic pNET whose disease had progressed during or after chemotherapy. Patients were treated with everolimus either as a single agent or in combination with standard monthly octreotide LAR. Everolimus monotherapy produced a modest response rate of 9.6% [all partial responses (PRs)] and stable disease (SD) in 67.8% of patients, whereas combination therapy yielded a 4.4% response rate (all PRs) and SD in 80% of patients. However, PFS responses were more promising: 9.7 months for patients receiving single-agent everolimus and 16.7 months for patients receiving the everolimus-octreotide LAR combination [13].
Everolimus has also been evaluated in two large, multicenter, randomized phase III studies of patients with NET. In RADIANT-2, everolimus plus octreotide LAR was compared with placebo plus octreotide LAR in 429 patients with functionally active, advanced, moderately or well-differentiated NET associated with the secretory symptoms (carcinoid syndrome) [14]. Patients treated with everolimus plus octreotide LAR had a median PFS of 16.4 months compared with 11.3 months for placebo plus octreotide LAR [hazard ratio (HR) 0.77; 95% confidence interval (CI) 0.59–1.00; p = 0.026; prespecified boundary p ≤ 0.0246] [14]. RADIANT-3 compared the efficacy of daily everolimus 10 mg versus placebo, both in conjunction with best supportive care, in 410 patients with advanced low- or intermediate-grade pNET [7]. Everolimus significantly increased median PFS by 6.4 months compared with placebo (11.0 vs. 4.6 months, respectively; HR 0.35; 95% CI 0.27–0.45; p < 0.001). Adverse events were predictable and manageable [7]. In May 2011, everolimus was approved by the FDA for the treatment of progressive pNET in patients with unresectable, locally advanced or metastatic disease. In September 2011, everolimus was approved in the European Union for the treatment of patients with unresectable progressive pNET.
The use of everolimus in combination with other agents also appears to be a promising therapeutic proposition. In a small phase I/II clinical trial of 24 patients, the safety and efficacy of everolimus plus temozolomide in patients with advanced pNET were evaluated [15]. On completion of a maximum of 6 treatment cycles, study participants without disease progression were continued on daily everolimus monotherapy. The combination of everolimus plus temozolomide was found to be promisingly effective against advanced pNET, with 35% of patients experiencing PR and 53% achieving SD. The most common grade 3 and 4 toxicities were anticipated hematologic effects.
Given the slowly progressing nature of well-differentiated NET, even in advanced cases, patients are likely to be treated with targeted agents including everolimus for many months and possibly years; therefore, issues of long-term safety and compliance will require special attention [16].
Multiple trials investigating the use of everolimus for the treatment of NET, either as monotherapy or in combination with various other agents, are ongoing. The various combination regimens under investigation include everolimus plus pasireotide, bevacizumab, erlotinib, cixutumumab, vatalanib and several cytotoxic agents. Some of these clinical studies have completed patient accrual, and results are anticipated in the near future.
VEGF Pathway
The important role of new blood vessel formation and growth in different pathologic processes, including tumorigenesis, is well established. VEGF, the principal growth factor responsible for angiogenesis, consists of a family of 6 proteins produced by different cell types. VEGF initiates the process of neovascularization through interaction with 3 specific transmembrane receptors that are expressed mostly on the surfaces of endothelial cells (fig. 2) [17]. Although the role of VEGF receptor 1 (VEGFR-1) is not well understood and VEGFR-3 may play a role in the mediation of lymphangiogenesis, VEGFR-2 is thought to be the most important in mediating tumor cell angiogenesis [17]. NET are characterized by high vascularity and overexpression of VEGF and at least 2 of its receptors (VEGFR-1 and -2) [18]. There are 2 groups of targeted agents that interact with the VEGF pathway: direct VEGF blockers such as bevacizumab, and multitargeted tyrosine kinase inhibitors (TKIs), which are directed against VEGFR and include sunitinib, pazopanib and sorafenib.

Role of the VEGFR tyrosine kinases in different cell types. VEGFR-1 and VEGFR-2 are expressed in the cell surfaces of most blood endothelial cells. VEGFR-3 is largely restricted to lymphatic endothelial cells. There is much evidence that VEGFR-2 is the major mediator of endothelial cell mitogenesis and survival as well as angiogenesis and microvascular permeability. In contrast, VEGFR-1 does not mediate an effective mitogenic signal in endothelial cells. Adapted with permission from Macmillan Publishers Ltd: Nat Med 2003;9:669–676. ©2003.
Role of the VEGFR tyrosine kinases in different cell types. VEGFR-1 and VEGFR-2 are expressed in the cell surfaces of most blood endothelial cells. VEGFR-3 is largely restricted to lymphatic endothelial cells. There is much evidence that VEGFR-2 is the major mediator of endothelial cell mitogenesis and survival as well as angiogenesis and microvascular permeability. In contrast, VEGFR-1 does not mediate an effective mitogenic signal in endothelial cells. Adapted with permission from Macmillan Publishers Ltd: Nat Med 2003;9:669–676. ©2003.
Bevacizumab
Bevacizumab is a humanized monoclonal antibody that binds to circulating VEGF-A. In 2004, it was the first FDA-approved inhibitor of angiogenesis and is indicated for treating several types of malignancies, including metastatic colorectal cancer, nonsquamous non–small cell lung cancer, glioblastoma multiforme and metastatic renal cell carcinoma [19].
The efficacy of bevacizumab in treating metastatic or unresectable carcinoid tumors was studied in 44 patients in a phase II randomized trial [20]. Patients were randomly assigned to receive bevacizumab plus octreotide LAR or pegylated interferon (IFN) alpha-2b plus octreotide LAR for 18 weeks. At disease progression or the end of 18 weeks (whichever occurred first), patients were treated with a combination of all 3 medications until disease progression. The results appeared promising: only 5% of patients initially treated with bevacizumab had disease progression compared with 32% treated with IFN alpha-2b. The effects of bevacizumab appeared to be a direct result of its impact on tumor blood circulation, reducing blood flow by 49% on day 2 of treatment; patients in the IFN arm did not show comparable effects. At the time of publication, 27 of 44 patients were alive; therefore, median PFS had not been reached. The reported 1-, 2- and 3-year survival rates were 93, 67 and 56%, respectively [20].
Combinations of bevacizumab plus other agents are also under investigation, with several phase II trials reporting promising data. In one study, the combination of bevacizumab plus temozolomide was shown to be effective in patients with advanced NET, with 27 of 29 patients (93%) demonstrating PR or SD [21]. In a small phase II study of 31 evaluable patients, a combined regimen of bevacizumab plus capecitabine and oxaliplatin resulted in PR in 23% and SD in 71% [22]. Of particular note, 6 of 7 patients with pNET had PR. Overall, the 1-year PFS with this treatment combination was 52% and median PFS was 13.7 months [22]. The combination of bevacizumab and FOLFOX (oxaliplatin, 5-fluorouracil and leucovorin) has also been evaluated in a small study of patients with NET [23]. Two of 6 patients with pNET had PR compared with 1 of 5 patients with small-bowel (carcinoid) NET, whereas SD was observed in the majority of patients regardless of primary site. A bevacizumab/everolimus combination has also demonstrated promising early results: in a small, randomized, run-in study of 39 patients with low- to intermediate-grade NET, 26% experienced PR and 67% had SD [24].
Other trials, planned and ongoing, aim to assess the role of bevacizumab in various combinations in patients with NET. One of the largest (estimated enrollment 400 patients) is the randomized Southwest Oncology Group phase III trial (SWOG S0518 trial; NCT00569127; www.clinicaltrials.gov) of bevacizumab plus octreotide LAR versus IFN-alpha plus octreotide LAR. In addition, as previously noted, a phase II study of bevacizumab plus the mTOR inhibitor temsirolimus in patients with recurrent or progressive pNET or carcinoid tumors is also under way (NCT01010126; www.clinicaltrials.gov).
Sunitinib
Sunitinib is a member of the family of multitargeted receptor TKIs. It inhibits all 3 types of VEGFR plus several other tyrosine kinase receptors [25]. In early studies, sunitinib demonstrated clinical efficacy in pNET. In a phase II study, patients with pNET had an overall response rate of 16.7% compared with 2.4% in patients with carcinoid tumors [26]. Results from a second phase II study suggested that sunitinib could delay time to tumor progression after hepatic artery embolization in patients with NET and liver metastases [27].
In a phase III, double-blind, placebo-controlled trial, sunitinib was compared with placebo in patients with advanced, well-differentiated pNET [6]. The study was initially designed to enroll 340 patients, but was discontinued prematurely after an early, unplanned analysis of 171 patients indicated an increased number of deaths and serious adverse events in the placebo group and increased PFS in patients treated with sunitinib [6]. Patients in the sunitinib group (37.5 mg/d) had a median PFS of 11.4 months compared with 5.5 months for the placebo group (HR 0.42; 95% CI 0.26–0.66; p < 0.001) [6]. The objective response rate was 9.3% in the sunitinib arm and 0% in the placebo arm (p = 0.007). Based on the results of this phase III study [6], sunitinib was approved by the European Medicines Agency (EMA) in November 2010 for the treatment of patients with unresectable or metastatic well-differentiated pNET with disease progression. In May 2011, the FDA approved sunitinib for the same indication after reanalyzing the data from the phase III trial and calculating a shorter median PFS of 10.2 months for patients in the sunitinib group and 5.4 months for patients on placebo (HR 0.43; 95% CI 0.27–0.67; p < 0.001) [28].
Pazopanib
Pazopanib is a small-molecule TKI, available in oral form, and was approved by the FDA in 2009 for the treatment of advanced renal cell carcinoma. Like sunitinib, pazopanib is believed to exert its effects largely through the inhibition of VEGFR and platelet-derived growth factor receptor (PDGFR). In a phase II clinical study, pazopanib plus octreotide LAR achieved a 17% response rate in patients with low-grade pNET, although no response was observed in patients with carcinoid tumors [29]. Median PFS times were 11.7 and 12.7 months for pNET and carcinoid tumors, respectively [29].
Sorafenib
Another representative of this family of multitargeted receptor TKIs, sorafenib, targets just one of the VEGF receptors (VEGFR-2), along with PDGFR-β and Raf kinase [30]. Sorafenib was approved in 2005 by the FDA for the management of advanced renal cell carcinoma and in 2007 by both the EMEA and FDA for the management of hepatocellular carcinoma.
In a phase II study, sorafenib as a single agent demonstrated modest activity against metastatic NET, with PR in 10% of patients with carcinoid tumors and in 10% with pNET [31]. Minor responses were observed in 12.9% of patients (6% in patients with carcinoid tumors and 20.9% with pNET). At 24 weeks, PFS was reported in 40% of evaluable patients with carcinoid tumors and 60.8% of evaluable patients with pNET. Of note, 43% of patients experienced significant toxicity (grade 3/4). The preliminary results of a more recent phase II trial evaluating the combination of sorafenib plus bevacizumab in patients with advanced NET demonstrated an overall response rate of 7.3% and a disease control rate of 95.1%, with a mean PFS of 12.1 months (95% CI, 10.6–13.6) [32].
Somatostatin Receptor Pathway
Somatostatin, normally produced in the brain, pancreas, stomach and intestine, is a hormone that targets transmembrane G-protein–coupled somatostatin receptors, which interact with multiple secondary hormones and facilitate the regulation of cell growth, neurotransmission processes and functioning of the endocrine system. There are 5 known types of somatostatin receptors: SST1, SST2, SST3, SST4 and SST5. Most (approx. 80%) carcinoid tumors express somatostatin receptors. For many years, somatostatin receptors were the only known specific target in the management of carcinoids until other pathways of carcinogenesis were discovered that led to the development of additional therapeutic agents. The use of SSAs in the treatment of patients with carcinoid tumors is a well-established practice [33].
Somatostatin Analogs
Octreotide/Octreotide LAR
Somatostatin is a peptide, the structure of which includes 14 amino acids; it has high affinity for all 5 types of somatostatin receptors. Its commercially available analog, octreotide, consists of 8 amino acids and binds with high affinity to SST2, with a lower affinity for SST5. Octreotide exhibits stronger inhibition of growth hormone, glucagon and insulin than somatostatin.
Octreotide is approved for the treatment of severe diarrhea and flushing in patients with carcinoid syndrome, profuse watery diarrhea related to vasoactive intestinal peptide-secreting tumors and acromegaly. A long-acting release form (octreotide LAR) that is administered as a monthly injection is now available.
Although the efficacy of octreotide in controlling the symptoms of carcinoid syndrome is well known, octreotide has not, until recently, been evaluated for a possible direct antitumor effect on NET. The PROMID study was a prospective, randomized, placebo-controlled, double-blind study in patients with low-grade metastatic NET of the midgut. It demonstrated that the use of long-acting octreotide more than doubled the time to progression compared with the placebo group (14.3 vs. 6 months). The level of functional activity of the tumor did not affect the response rate. Clinically, SD was the most frequently observed response, and patients with a relatively low tumor burden seemed to benefit the most [3].
Lanreotide
Lanreotide is another synthetic SSA that binds to the same receptors to which octreotide binds. However, it has a higher affinity for peripheral receptors and a significantly longer half-life. In an open, multicenter, cross-over study, lanreotide was shown to have efficacy equal to that of octreotide in controlling flushing and diarrhea and in reducing levels of plasma serotonin and urinary 5-hydroxyindoleacetic acid [34].
Pasireotide
Pasireotide is an SSA with a particularly high binding affinity for SST5 (40-fold), SST1 (30-fold) and SST3 (5-fold) compared with octreotide, but with the same affinity for SST2 [35].
It is a twice-daily medication administered subcutaneously with dose escalation until symptoms are controlled. It was shown in a phase II study to be effective in controlling symptoms in 27% of the patients with advanced carcinoid tumors whose treatment with octreotide LAR had failed [36].
A phase III study comparing the long-acting formulations pasireotide LAR and octreotide LAR in patients with advanced NET is recruiting participants (NCT00690430). Other studies are evaluating the combination of pasireotide with other agents, including everolimus.
Peptide Receptor Radionuclide Therapy
Targeting NET with SSAs conjugated with radioisotopes is an attractive therapeutic option given the high expression of somatostatin receptors on the surfaces of neuroendocrine tumor cells. By using radiolabeled SSAs, the radioactive particles can be delivered more directly to the tumor cells.
Several variants of such conjugates have been developed, with yttrium-90 (90Y), lutetium-177 (177Lu) and indium-111 (111In) evaluated the most comprehensively [37]. A complete description of the different radionuclides, peptides and chelators is outside the scope of this review.
The earliest studies of peptide receptor radionuclide therapy (PRRT) used 111In as the radionuclide, but the characteristics of 111In were not optimal for the management of NET and it is rarely used now. The most commonly used isotopes are 90Y and 177Lu, but no randomized trials have been performed comparing those two radionuclides, and no particular isotope has emerged as superior and preferred. Cross-trial comparison of these radionuclides is difficult given the different methodology used for the different studies. Both lutetium and yttrium continue to be widely used, especially in Europe. The reported radiographic response rates range from 4 to 35%, and radiographic response has been correlated with improved survival [37].
The use of radiolabeled SSAs is of great theoretical and practical research interest. This method is being used in multiple European medical centers for the treatment of patients with NET. However, the exact role of this treatment in the management of NET remains to be defined, and well-designed trials comparing PRRT with medical therapy are needed. One such trial comparing PRRT with high-dose SSA therapy is being launched and is expected to shed light on the role of PRRT in the modern management of neuroendocrine malignancies. Overall, PRRT is well tolerated, seems to significantly slow progression and has relatively few serious adverse events. The most common severe adverse event is renal insufficiency, which may be irreversible. Myelodysplastic syndrome has also been reported after PRRT but seems to be uncommon. PRRT has not been approved by the FDA for use in the USA and is not readily available [37].
EGFR Pathway
Epidermal growth factor receptor (EGFR), a transmembrane tyrosine kinase receptor, is activated when a ligand (EGF or related factors) binds to its extracellular domain [38]. Activation of the EGFR leads to its dimerization, triggering intracellular signaling cascades [38]. Three major signaling pathways are recognized as downstream mediators of EGFR effects: (1) the Ras/Raf/MEK/ERK pathway, involved in proliferation, tumor invasion and metastatic spread; (2) the PI3K-Akt pathway, which regulates the activation of nuclear transcription factors and thus affects the major signals involved in apoptosis and cellular survival; and (3) the JAK/STAT pathway, which plays a role in activating the transcription of genes important for cell survival (fig. 3) [38].
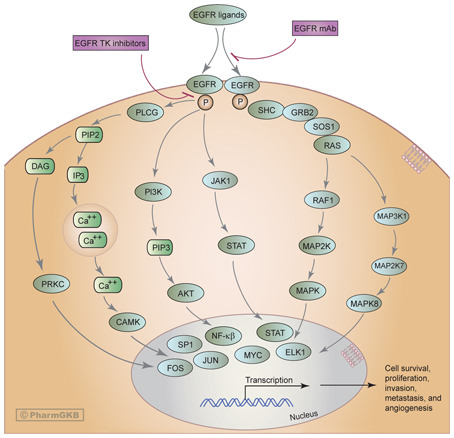
Monoclonal antibodies and inhibitors of EGFR-specific tyrosine kinase may affect numerous genes and pathways. Arrows represent activation, bars represent inhibition. Reprinted with permission from PharmGKB and Stanford University. © PharmGKB. http://www.pharmgkb.org/do/serve?objCls=Pathway&objId=PA162356267.
Monoclonal antibodies and inhibitors of EGFR-specific tyrosine kinase may affect numerous genes and pathways. Arrows represent activation, bars represent inhibition. Reprinted with permission from PharmGKB and Stanford University. © PharmGKB. http://www.pharmgkb.org/do/serve?objCls=Pathway&objId=PA162356267.
Gefitinib
Gefitinib is a targeted agent that selectively inhibits receptor tyrosine kinases, including EGFR. In a phase II study of patients with advanced metastatic NET, gefitinib exhibited somewhat promising initial results [39]. At 6 months, 61% of patients with carcinoid tumors and 31% with pNET were progression-free; however, objective responses for each group were low (5 and 9.6%, respectively) [39].
IGF-1R Pathway
IGF-1R is a transmembrane receptor tyrosine kinase activated by IGF-1 and IGF-2. After ligand binding and activation, IGF-1R signals are transmitted through components of the PI3K/Akt/mTOR and the Ras/Raf/MEK/ERK pathways, inducing cellular proliferation and upregulating antiapoptotic activity (fig. 4) [40]. It has been shown that various receptor tyrosine kinases, including IGF-1R, are overexpressed in NET, making this another potential target for therapy [41].
![Fig. 4. IGF-1 signaling pathway. Arrows represent activation, bars represent inhibition. Activation of the IGF-1R by IGF ligands results in enhanced proliferation and strong survival signals in tumor cells. Although complex, the signaling pathways involved are primarily the Ras-Raf-mitogen-activated protein kinase (MAPK)-ERK proliferation pathway and the PI3-PKB-Akt survival pathway. Adapted with permission from [40].](https://karger.silverchair-cdn.com/karger/content_public/journal/ocl/83/3/10.1159_000339539/2/m_000339539_f04.jpeg?Expires=1716365240&Signature=zedd20UjjGBY~dtP6ZldLfux8GGXYyM7Z-16HY4HebhLMUqZVcs33daZp0yoChz20-JIS9m4mrgPA7R9gZqCI3aRBPyZHNvHyoO~L5ylCtSeWBbAQua940pjCOsCCXGfug090VV6hUtZEHnPnSDrpWff-pgZsUuRU6RxIWnBgq8eQcd6ePLk8ay6puOsY8Vh3b7qEZZaPPk~hzLdwWpPnYM1ic~JYuiNIseSjkTWVlL2K5NkMMeeGf2bPuK6MBhR8QEBf7Jn-ZBbUsyspoZvdU57ZYk8ScOELB00O8P2TTDBIVSa3KXyJvRONaxVvJ72KeEkRldHmGF6RZM6SaLEpw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
IGF-1 signaling pathway. Arrows represent activation, bars represent inhibition. Activation of the IGF-1R by IGF ligands results in enhanced proliferation and strong survival signals in tumor cells. Although complex, the signaling pathways involved are primarily the Ras-Raf-mitogen-activated protein kinase (MAPK)-ERK proliferation pathway and the PI3-PKB-Akt survival pathway. Adapted with permission from [40].
IGF-1 signaling pathway. Arrows represent activation, bars represent inhibition. Activation of the IGF-1R by IGF ligands results in enhanced proliferation and strong survival signals in tumor cells. Although complex, the signaling pathways involved are primarily the Ras-Raf-mitogen-activated protein kinase (MAPK)-ERK proliferation pathway and the PI3-PKB-Akt survival pathway. Adapted with permission from [40].
AMG479 (ganitumab)
AMG479 (ganitumab) is a fully human monoclonal antibody against IGF-1R undergoing evaluation in clinical trials. AMG479 prevents the binding of IGF-1 and IGF-2 to IGF-1R [42]. In a phase I trial that included 5 patients with previously treated metastatic NET, AMG479 demonstrated promising activity (1 PR and 4 SD) and good tolerability [43].
Cixutumumab
Cixutumumab, another fully human immunoglobulin G1 monoclonal antibody directed against IGF-1R, is also in the early stages of clinical development. A phase II study that aims to evaluate the combination regimen of cixutumumab plus octreotide LAR in patients with progressing metastatic midgut carcinoid tumors and pNET is actively accruing patients [44]. In addition, the combination of cixutumumab, everolimus and octreotide is being studied in patients with advanced NET in a phase I trial (NCT01204476; www.clinicaltrials.gov).
Histone Deacetylase Pathway
Histone deacetylases (HDACs) are a family of intracellular enzymes that are involved in the removal of acetyl groups from ε-N-acetyl lysine amino acids on histones and play important roles in the process of DNA transcription and regulation of gene expression. Inhibitors of HDAC (e.g. valproic acid) have been used in neurology for many years as antiepileptics and in psychiatric practice as mood stabilizers. Recently, HDAC inhibitors were shown to be effective in the treatment of certain types of cancer [45], and although their exact mechanism of action in cancer therapy is not clear, 2 agents from this group have been approved by the FDA for the treatment of cutaneous T-cell lymphoma (vorinostat in 2006 and romidepsin in 2009). However, the level of adverse events and toxicity with HDAC is a cause for concern. Vorinostat has been investigated for efficacy in a variety of solid tumor sites, but studies showed a large number of adverse events as well as a high rate of discontinuations, that made accurate efficacy evaluations problematic [45]. Similarly, romidepsin (FR901228) has been studied in patients with neuroendocrine malignancies but was found to have unacceptable cardiotoxicity [46]. A phase I trial of romidepsin (at lower doses than used in previous studies) in combination with gemcitabine in patients with pancreatic and other advanced solid tumors also showed dose-limiting toxicities; however, 5 of 9 patients with pancreatic cancer experienced SD for ≥4 cycles. This combination is being further explored in phase II studies [47].
Protein Degradation Pathways
Proteasome Inhibitors
Proteasomes are large multicatalytic protein complexes responsible for the degradation of most intracellular proteins [48]. This pathway is involved in a number of cellular processes crucial to oncogenesis. In vitro, inhibitors of proteasomes have demonstrated the ability to induce apoptosis in cancer cell cultures. Bortezomib is a potent and selective inhibitor of the proteasome. It was the first commercially available proteasome inhibitor and is approved by the FDA for the treatment of patients with relapsed multiple myeloma and mantle-cell lymphoma. Recent data indicate that bortezomib may also exert some of its anticancer effect through the PI3K/Akt/TOR pathway, and an in vitro study suggested that it has some ability to induce apoptosis in NET cell lines [49]. However, when the activity of bortezomib against metastatic NET was evaluated in a phase II clinical trial, the results were discouraging, with no responses observed [50]; there are no ongoing clinical trials evaluating the use of bortezomib in NET.
Immunomodulating Therapy
Thalidomide
Thalidomide, an agent used for the management of multiple myeloma, has an incompletely understood mechanism of action. Although it is known to be an inhibitor of the tumor necrosis factor-α pathway, it has also been shown that thalidomide has the ability to interfere with the VEGF and basic fibroblast growth factor pathways, thus inhibiting the development of new blood vessels [51]. The possible efficacy of thalidomide in the management of NET was suggested by a phase II study in which 81% of patients with advanced NET treated with the drug had disease stabilization at 12 weeks, and 78% had disease stabilization after 24 weeks [52]. In a second phase II trial in 18 patients with metastatic NET, thalidomide as a single agent achieved SD in 69% of patients, but no objective responses were observed [53]. Another recent phase II study evaluated the combination of thalidomide plus temozolomide in patients with pNET (n = 11), carcinoid tumors (n = 15) and pheochromocytomas (n = 3) [54]. This combination achieved a radiologic response rate for 45% of patients with pNET (including one complete response) and 7% with carcinoid tumors. Median duration of response for the entire group was 13.5 months, 1-year overall survival was 79% and 2-year overall survival was 61%. However, it is not known how much each individual therapeutic component contributed to the overall activity [54]. Despite these positive preliminary data in small patient populations, no open trials are addressing the use of thalidomide in NET.
c-kit and PDGFR Pathways
Imatinib
Imatinib is an oral TKI that specifically targets enzymes in several cellular pathways, including abl (which plays a prominent role in chronic myelogenous leukemia), c-kit and PDGFR pathways. In vitro activity of imatinib has been shown against several human NET cell lines. A phase II clinical trial with imatinib demonstrated modest clinical activity against advanced neuroendocrine carcinomas (3.7% PR and 63% SD) [55]. Biochemical response to treatment was a prognostic factor of longer PFS (115 vs. 24 weeks; p = 0.003), as was concurrent octreotide therapy (49 weeks with octreotide vs. 14 weeks without octreotide; p = 0.03) [55].
Conclusions
First-line therapy for patients with NET has long included SSA, but new therapies are required for patients with progressive disease. Targeted therapies appear to be effective treatment options for patients with advanced GEP-NET. Multiple molecular pathways of cellular proliferation have been identified, and several targets within these pathways have been explored. Everolimus (an mTOR inhibitor) and sunitinib (a multitarget TKI) were recently shown to substantially prolong PFS in patients with pNET. Both drugs are now approved by the FDA and the EMA for treating patients with advanced pNET. However, the lack of available comparative studies between these targeted therapies makes it difficult to suggest the optimal sequence of treatments. Other targeted agents are under clinical investigation and are likely to play prominent roles in the management of neuroendocrine malignancies in the future. Hopefully, ongoing trials will help guide clinicians in selecting the most appropriate clinical strategy.
Disclosure Statement
B.G.N. and T.R.H. have nothing to disclose.
J.R.S. serves on the advisory boards for Pfizer and Novartis and on the speakers bureau for Genentech.

