

Abstract
Background: In default of a plausible and satisfactory causal treatment for hemorrhagic stroke, a role of matrix metalloproteinases (MMPs) in the pathogenesis of cerebrovascular diseases has recently been widely discussed. The well-known impact of MMPs on extracellular matrix destruction triggered by inflammation as a foundation for several diseases, including stroke, is very much in evidence. Newly, some additional aspects of MMP function considering their intracellular activity crucial for neuronal death following ischemic brain damage have emerged. The effect of blood-brain barrier disruption caused by MMPs on the prognosis in patients suffering from spontaneous intracerebral hemorrhage (ICH) has been of interest since it throws a new light upon the pathogenesis, course and possible therapeutic approaches for this least treatable and at the same time most life-threatening form of stroke. Hence, we primarily aimed to review the current clinical knowledge on the significance of metalloproteinase activation in the course of spontaneous intracranial hemorrhage in humans. We also provide a brief characterization of the MMP enzyme family and report on the latest findings on issues arising from experimental studies. Methods: A Medline search using the following key words was performed: matrix metalloproteinases + spontaneous intracerebral hemorrhage/intracranial hemorrhage/bleeding/hemorrhagic stroke. We accepted studies reporting on MMP expression in adult patients with spontaneous ICH, as well as its relation to radiological and clinical features and patients’ outcome. For the final review, 18 clinical studies were considered. MMP inhibition was reviewed on the basis of 11 relevant experimental studies. Also, some relevant reports on the biology of MMPs and their pathophysiology in ICH were reviewed. Results and Conclusions: Many studies provide convincing evidence of a detrimental role of MMPs in ICH, stressing their association with neuroinflammation. The role of MMPs in hemorrhagic stroke appears critical for hematoma and brain edema growth as well as for neuronal death, which are understood as secondary brain injury and may have a considerable clinical impact. Although data on human spontaneous ICH are scarce and mostly based on small populations, they reveal the apparent correlation between MMPs and clinical and radiological ICH features as well as the functional outcome, which might rationalize future therapeutic strategies. However, attempts at MMP inhibition in spontaneous ICH have solely been made under experimental conditions and were associated with a wide range of possible side effects. Therefore, further comprehensive, elucidating investigations in this field are vital before any conclusions could be translated to humans.
Introduction: New Challenges in the Pathogenesis of Spontaneous Intracerebral Hemorrhage
Cerebrovascular research, as yet sparingly focused on hemorrhagic stroke, unlike the case of ischemic insults, has not established any milestones in the therapy of this severe condition. Furthermore, it seems to prove the unique and very complex nature of this type of stroke. This section is intended to provide a brief summary regarding recent interest in the course of spontaneous intracerebral hemorrhage (ICH).
Spontaneous ICH accounts for about 15% of all stroke cases, and up to 50% of patients with this condition die within 1 month after onset [1]. Age, intraventricular bleeding extension, baseline hematoma size and Glasgow Coma Scale score but first and foremost hematoma growth within the first 24 h as crucial for the prognosis in spontaneous ICH patients were recently intensively studied with the hope of achieving a better understanding of the pathogenesis of spontaneous ICH [2,3]. The Factor Seven for Acute Hemorrhagic Stroke Trial failed to confirm clinical benefit of applying recombinant factor VIIa as early treatment of spontaneous ICH [4]. Although significant hematoma growth reduction was shown after recombinant factor VIIa administration, it has been pointed out that inadequate patient selection could have counterbalanced the primarily promising effects [5,6].
Nevertheless, bleeding growth remains an important target when considering new therapeutic approaches.
When discussing the phenomenon of bleeding formation and enlargement after ICH, the significance of perihematomal edema (PHE) needs to be raised. Xi et al. [7] hypothesized edema formation to be vital in secondary brain injury after spontaneous ICH onset, contributing to blood-brain barrier (BBB) damage and hematoma enlargement. PHE formation was shown to start as soon as 3 h after spontaneous ICH onset and to increase even up to 14 days [8,9]. Gebel et al. [10] proved relative PHE (PHE volume divided by hematoma volume measured on baseline brain CT scans) to be an independent predictor of poor neurological outcome but not mortality 3 months after spontaneous ICH onset. However, the pivotal role in edema formation has been assigned to the thrombolytic cascade and production of thrombin within 24 h. It is worth noting that expression of prothrombin mRNA additionally by neurons, without opening of the BBB, can amplify that effect by directly promoting the development of edema and enhancing inflammation [11]. Thrombin may also induce brain edema development by influencing the c-Jun N-terminal kinase pathway. Michel-Monigadon et al. [11] showed that c-Jun N-terminal kinase inhibition reduced brain edema after experimental brain hemorrhage.
Thus, the origin of primary PHE after spontaneous ICH is vasogenic and not cytotoxic, as it develops due to an increase in the permeability of the BBB after the onset of ICH and is triggered by proinflammatory agents [7].
Matrix Metalloproteinases: Profile and Properties
Matrix metalloproteinases (MMPs) together with other proteases such as serine or cysteine proteases belong to the group of endopeptidases. They play a unique role in remodeling of extracellular matrix (ECM) due to their ability to splinter its integrity. This makes them important contributors in many physiological processes including blastocyst implantation, embryonic development, nerve growth, endometrial cycling, mammary gland morphogenesis, bone remodeling, wound healing, angiogenesis and apoptosis. Also of the utmost importance is their participation in a number of pathologic processes; MMPs are ubiquitously employed, for example, in cancer invasion and metastasis. Moreover, they are assumed to induce tissue destruction in, for example, aortic aneurysm, rheumatoid arthritis, gastric ulcer, dilated cardiomyopathy and fibrotic lung disease as well as in many diseases of the central nervous system [12,13,14]. They also promote anoikis – cell death due to homelessness – as a result of matrix disintegration [14]. Intracellular MMP function has been widely studied, providing new insight into the role of this enzyme family. MMPs have been shown to invade cells in the process of endocytosis and to take part in protein cleavage in the cytosol and in some of the organelles [15]. More importantly, as reviewed by Mannello and Medda [16] , some MMPs can enter the cellular nucleus and influence gene transcription and DNA repair, leading to decreased function of poly-ADP-ribose polymerase-1 (PARP-1) and X-ray repair cross-complementing protein-1, which are responsible for the process of DNA reparation. This in turn results in apoptosis.
MMPs along with seralysins, astacins and adamalysins or a disintegrin and metalloproteinase (ADAMs)/ADAMs with thrombospondin motifs are also known as metizins because of the specific structure of their catalytic domain containing a zinc atom [13].
In fact, there are three functional domains that form the MMPs structure and which determine the idiosyncrasy of the MMPs, merging these 24 enzymes into one superfamily of zinc peptidases. The basic constellation comprising a signaling peptide (predomain), propeptide (prodomain) and catalytic domain is identical to the smallest MMP, called matrilysin (MMP-1). With the broadening of knowledge regarding MMPs, the terminology used to name these enzymes has been changing. Nevertheless, the system based on substrate specificity is still widely used. It comprises 8 subgroups named after their substrates: matrilysins, collagenases, stromelysins, gelatinases, convertase-activatable MMPs, membrane-associated MMPs, MMP-23A and MMP-23B (fig. 1). Altogether, MMPs are responsible for entire ECM remodeling [13].
![Fig. 1. Structural division of MMPs based on Overall and López-Otín [13]. The diagram represents 8 independent subgroups of MMPs, which are all based on the minimal domain set. The hemopexin domain differs slightly among the subgroups, thus determining the substrate specificity. Additional inserts and domains provide further specificity differentiations between the subgroups (graphic by M. Florczak-Rzepka and A. Sułkowska).](https://karger.silverchair-cdn.com/karger/content_public/journal/ced/34/4/10.1159_000341686/2/m_000341686_f01.gif?Expires=1716304580&Signature=paMVqciy-6ReZvPw6CvQZ1leeFanZppiouR-ev00gGsIBH-LYTDT27btzgvMTPv8r5-fwoSklAdVbIo6FZ9nJUGSVkGtxZVRCemLjwGkceTlaqoIT6tES~nSpJ5nC2NrOhRiGKZBh147KlerUniDRJHxlw~uW0Mfgdk6RIBLxvoSs4pKcjpoIdoPvq7W0QAbil~ZCwdcsG5JI7ohAjvDn-qNoJZaJkvmcLtHkFiAFM8dlg1-XC6hjDMoZkGIZ31ZBjsGR8bTYCacGHH1g8bY40-94Bmd3IDmtEsDz2ufJzDjMWnQyiJdMAT-phleQJevrGlE9PDB-4~1dWvjvuboyA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Structural division of MMPs based on Overall and López-Otín [13]. The diagram represents 8 independent subgroups of MMPs, which are all based on the minimal domain set. The hemopexin domain differs slightly among the subgroups, thus determining the substrate specificity. Additional inserts and domains provide further specificity differentiations between the subgroups (graphic by M. Florczak-Rzepka and A. Sułkowska).
Structural division of MMPs based on Overall and López-Otín [13]. The diagram represents 8 independent subgroups of MMPs, which are all based on the minimal domain set. The hemopexin domain differs slightly among the subgroups, thus determining the substrate specificity. Additional inserts and domains provide further specificity differentiations between the subgroups (graphic by M. Florczak-Rzepka and A. Sułkowska).
Under normal quiescent conditions, MMPs expression is very limited and MMPs are detected either in small amounts or not at all in tissues or in the circulation.
Usually MMPs are synthetized as nonactive, latent forms called zymogens. Their latency is maintained by preserving the pre-/prodomain structure. This is achieved by coordination between the Zn2+ ion in the catalytic domain and the SH group of unpaired cysteine in the highly conserved sequence in the prodomain (fig. 2) [17].

Autocatalysis in the process of MMPs activation. Prodomain cleavage may occur either extracellularly or inside cells as a result of several processes. The best known is direct proteolysis mediated by serine proteases or other active MMPs. Free oxygen or nitrogen radicals (reactive oxygen species/reactive nitrogen species) may cause disruption of the Cys-Zn interaction. Allosteric activation causes distortion of the catalytic site in MMPs. All reactions are followed by prodomain cleavage, which definitely leads to MMP activation. All depicted pathways occur extracellularly. Proteolysis and activation by reactive oxygen species/reactive nitrogen species can also occur inside cells. More details on MMP activation are to be found in the text (graphic by M. Florczak-Rzepka and A. Sułkowska).
Autocatalysis in the process of MMPs activation. Prodomain cleavage may occur either extracellularly or inside cells as a result of several processes. The best known is direct proteolysis mediated by serine proteases or other active MMPs. Free oxygen or nitrogen radicals (reactive oxygen species/reactive nitrogen species) may cause disruption of the Cys-Zn interaction. Allosteric activation causes distortion of the catalytic site in MMPs. All reactions are followed by prodomain cleavage, which definitely leads to MMP activation. All depicted pathways occur extracellularly. Proteolysis and activation by reactive oxygen species/reactive nitrogen species can also occur inside cells. More details on MMP activation are to be found in the text (graphic by M. Florczak-Rzepka and A. Sułkowska).
Importantly, the activation of MMPs is regulated on 3 main levels, starting with transcription, through activation of latent forms and ending up at the inhibition level [14]. The next three sections are devoted to these processes.
MMP Activation
Following signal peptide removal in the process of translation, usually inactive MMPs are secreted into the extracellular space to be activated there (fig. 3). However, recent studies on intracellular MMP activity provide evidence that the MMP activatory process may also take place inside the cell [15,16].
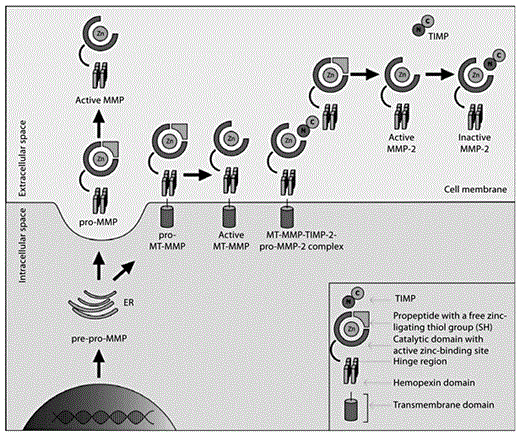
MMP activation pathway. Firstly, pre-pro-MMPs are expressed and transported through the endoplasmatic reticulum (ER) to the cell surface as zymogens devoid of the predomain. Most of the MMPs are secreted, and their activation takes place in the extracellular space. MMPs equipped with a transmembrane domain anchor to the cell membrane and are activated on the cell surface. Pro-MMP-2 binds to the complex molecule consisting of an active MT-MMP (transmembrane MMP) and TIMP to form the active MMP-2 form. Further information is to be found in the text (graphic by M. Florczak-Rzepka and A. Sułkowska).
MMP activation pathway. Firstly, pre-pro-MMPs are expressed and transported through the endoplasmatic reticulum (ER) to the cell surface as zymogens devoid of the predomain. Most of the MMPs are secreted, and their activation takes place in the extracellular space. MMPs equipped with a transmembrane domain anchor to the cell membrane and are activated on the cell surface. Pro-MMP-2 binds to the complex molecule consisting of an active MT-MMP (transmembrane MMP) and TIMP to form the active MMP-2 form. Further information is to be found in the text (graphic by M. Florczak-Rzepka and A. Sułkowska).
Crucial for enzymatic activation of MMPs is cleavage of the cysteine-zinc bridge (6- to 10-kDa N-terminal propeptide cleavage) in the process called ‘cysteine switch’. This leads to exposure of the zinc atom to water molecules and the thiol group to a substrate [17]. Afterwards, the prodomain is cleaved in a process called autocatalysis. Thus, the latent MMP form – proform or zymogen – becomes an active, lower-molecular weight MMP. There are at least 3 mechanisms by which the Cys-Zn coordination is broken, as reviewed by Cauwe and Opdenakker [15]. The main MMP activatory pathways are schematically depicted in figures 2 and 3 and described below:
(1) The first of these is proteolytic cleavage, which depends on catalytic site opening by proteases. The special role of serine proteases in this process may be explained by their involvement in many intra- and extracellular processes, such as the coagulation cascade. Plasmin, which belongs to the serine protease family, localizes in the vicinity of fibrin deposition or on mobilized cells, which allows it to take part in diverse processes such as fibrinolysis and ECM decomposition during cell migration. This specific localization is guaranteed by the proenzyme form, plasminogen, but firstly by the close proximity to plasminogen activator, which in turn depends on the type of plasminogen activator; tissue plasminogen activator anchors fibrin, whereas urokinase plasminogen activator sticks to membrane receptors [18].
Intracellular proteolytic activation depends on furin, one of the serine peptidases that was found to cleave some MMPs, like MMP-11 and membrane-type 1 (MT1)-MMP in the trans-Golgi apparatus network [19,20].
The link between serine proteinases and MMP activation builds up a complicated system of interactions and was therefore named the ‘protease web’ or ‘internet’, whereby active MMPs activate other MMPs [21].
An example of such a relationship is the MMP-2 activatory pathway, which is promoted by active MT1-MMP. This also applies to gelatinase B (MMP-9) and MMP-13. Interestingly, in the process of MMP-2 liberation, binding to tissue inhibitor of MMP (TIMP)-2 is required [12,14,22 ](fig. 3).
(2) Any cell injury may lead to an imbalance between the production and disposal of reactive oxygen species or reactive nitrogen species, causing oxidative or nitrosative stress, respectively. Oxygen or nitrogen free radicals may interact with the thiol group in the prodomain and expose catalytic zinc to bind with water molecules and so activate the MMP. An interesting fact is that a low concentration of some free radicals may activate some zymogens, such as pro-MMP-7 [23], but at the same time, prolonged exposure to free radicals and their persistent high concentration may, in contrast, inactivate some MMPs. Eventually, it was shown that in some cases there is no ultimate need for prodomain cleavage to achieve an active state of MMPs in the presence of free radicals. Therefore, the MMP proform is no more equivalent to the latent form or zymogen. Similar modification of the thiol group may also occur in the presence of nonphysiological agents, like heavy metal ions or alkylating reagents [15].
(3) Allosteric activation of MMPs consists of shape modification of the catalytic site in the MMP molecule. The same effect may be achieved by attaching a substrate, mainly nonphysiological agents, to the thiol group and disruption of the Cys-Zn coordination.
All the activatory pathways take place in the extracellular space [15].
The intracellular MMP activation process, as reviewed by Cauwe and Opdenakker [15], includes proteolysis and oxidative/nitrosative stress but also phosphorylation mediated by alkaline phosphatase, which was shown in the case of MMP-2 activation. Most frequently this process occurs in MMPs containing a fibronectin-like domain, but it has also been shown in the case of MT1-MMP in its cytoplasmatic domain.
Alternatively to prodomain cleavage, it was found that low-molecular weight MMPs lacking signal peptide and propeptide may be expressed in a process called alternative splicing, as in the case of MMP-11 [15].
Gene Expression
A wide range of factors can induce MMPs transcription. Some data based on cancer research indicate the role of growth factors, cytokines and oncogene products. Typically they are released by the matrix or infiltrating host defense cells but also by tumor cells themselves. Additionally, every change in the cell vicinity such as form alterations of the cell or mechanical stress can also trigger MMPs transcription. As yet, there is no single agent that has been found to be exclusively responsible for activating MMPs gene expression [13]. Nuclear factor (NF)-ĸB and other nuclear factors, as well as tumor necrosis factor-α (TNF-α), interleukin (IL)-1β and IL-6, are also constantly involved in the MMPs expression process [13,24] (fig. 4). By virtue of the variability in the promoter of MMP genes, another level of MMP diversity opens. Single-nucleotide polymorphisms may modify the processing of mRNA and consequently the activity of expressed proteases. It has been reported that a 736-base pair region in the promoter sequence of the gene encoding MMP-9 containing a cytosine-adenosine repeat may vary in length from 13 to up to 23 repeats in the human population and is associated with susceptibility to intracranial aneurysms [25]. In fact, one should consider that a polymorphism is merely a variation of the normal sequence, and hence, small-sample studies may not have enough power to demonstrate any phenotypic effect of such a genetic variant.

MMP regulation pathway. Selection of the most frequently involved factors and the scope of MMPs. uPA = Urokinase plasminogen activator; ROS = reactive oxygen species; SNP = single-nucleotide polymorphism; TGF-β = transforming growth factor β.
MMP regulation pathway. Selection of the most frequently involved factors and the scope of MMPs. uPA = Urokinase plasminogen activator; ROS = reactive oxygen species; SNP = single-nucleotide polymorphism; TGF-β = transforming growth factor β.
MMP Inhibition
Attenuation of MMPs activity is crucial to counterbalance their proteolytic extra- and intracellular activity [17]. There are several ways to inhibit MMPs, firstly by targeting factors that may possibly induce MMPs gene expression and secondly by influencing MMPs activity. In fact, there are two physiological MMP inhibitors (MMPIs). Outside tissues, in the plasma, α2-macroglobulin binds to MMPs, creating an irreversible complex. Inside tissues, specific TIMPs were found to be mainly responsible for regulation of MMPs activity [16].
Inhibition is achieved through formation of tight 1:1 noncovalent complexes with TIMPs. Four types of these natural inhibitors have been described so far, and each one is able to interact with any of the MMPs. However, there is a clear proclivity towards some combinations between MMPs and their inhibitors, such as TIMP-1-MMP-2 and TIMP-2-MMP-9 [17]. It is also worth noticing at this point that in the case of TIMP-2 and -3, lower concentrations can enhance rather than inhibit MMP-2. Inversely, at higher concentrations, MT1-MMP is entirely blocked, which results in a purely inhibitory effect on MMP-2 [18,26] (fig. 3).
However, the role of TIMPs is complex and extends beyond a strictly inhibitory effect on MMPs, at least in the case of cerebral ischemia [27,28].
What Is the Link between the Function of MMPs and the Pathogenesis of ICH?
ICH is a massive event, destroying brain tissues by induction of several mechanisms. Primary brain injury occurs within minutes after ICH onset and is related to the extravasated blood mass itself but also to the cell debris and plasma proteins. As described by Aronowski and Zhao [29], erythrocytes are lysed within 24 h, releasing hemoglobin along with its derivatives, including heme and iron, which are both capable of evoking oxidative cell damage by generating free radicals. It seems that the protective role of haptoglobin in building complexes with free hemoglobin, thus preventing its toxicity, is insufficient, as its recovery process, mostly by the liver, lasts 5–7 days. However, there is some evidence that haptoglobin may be produced at the site of bleeding. Nevertheless, it was also reported by the same group that in the course of ICH, endogenous antioxidative mechanisms may be depressed, such as that mediated by Nrf2. This was confirmed using Nrf2-knockout mice, which had much more extensive brain tissue injury than wild types after induced ICH [30]. Fortunately, peroxisome proliferator-activated receptor-γ-induced antioxidative enzyme (catalase) production was shown to be upregulated in experimental ICH [31,32].
The second part of the posthemorrhagic brain injury occurs in parallel and starts within hours from the ictus. Mediated by adhesion molecules, leukocyte infiltration as well as activation of other immune cells (macrophages, microglia, astroglia) results in cytokines (e.g. IL-1β, TNF-α) and chemokines overexpression. The main inflammatory pathway signaling molecule is thought to be NF-ĸB, which affects gene transcription of the above-mentioned key adhesion molecules, cytokines and chemokines as well as MMPs (fig. 4) [33,34,35,36]. Secondary brain injury results in disruption of the BBB, brain edema and massive neuronal death. The role of MMPs in both processes seems to be fundamental.
In the central nervous system, the basement membrane (BM) has a pivotal role for preserving the function of the BBB, which is made up of the following types of cells: perivascular endfeet of astrocytes, glia (microglia), pericytes, neuronal processes and the basal lamina (BM). The latter consists mainly of type IV collagen, laminin, fibronectin and elastin (fig. 5) [14,18,37].
![Fig. 5. BBB destruction by MMPs during the inflammatory process based on Abbott et al. [37]. The process includes fibrinolytic cascade, oxidative stress, neutrophil infiltration and activation of immune cells, e.g. microglia. All involved cells along with stimulated endothelium release cytokines and chemokines as well as MMPs. Invasion into the brain is followed by destruction of TJ proteins and BM decomposition caused by activated MMPs. t-PA = Tissue plasminogen activator. Further information is provided in the text (graphic by M. Florczak-Rzepka and A. Sułkowska).](https://karger.silverchair-cdn.com/karger/content_public/journal/ced/34/4/10.1159_000341686/2/m_000341686_f05.gif?Expires=1716304580&Signature=FwCTNiPsvPO1oJY-L9~z-DXC64rrx4v~6ysAMw4BVxbQEGz~ddEe90gZWBVA~0M5AYFP-giyXqXMbzIc8LZOZfY59jG69k7UjsVVQ0YLotVuMMA9gbAG0PB8Sd7m0PoP9QQvSgnOf4myM468LaucVRHycdwWw5BUBQqjTUIHlfsjChmfZz1xLEum7kgCob2ccB6zk13DPqhI0BaVEsMkD4mgSJq-5fEvRJSQZ-jhTrCqf4e5QC8CBoHCS9ZLnVDhhbfk2YeMemTpQjsCN1t7f1bDy0N4zgnU0HExKevRZL5R1kepM6bYKPJWu0mGTRpEzluXC0RV9Xv2DRSnarGcQw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
BBB destruction by MMPs during the inflammatory process based on Abbott et al. [37]. The process includes fibrinolytic cascade, oxidative stress, neutrophil infiltration and activation of immune cells, e.g. microglia. All involved cells along with stimulated endothelium release cytokines and chemokines as well as MMPs. Invasion into the brain is followed by destruction of TJ proteins and BM decomposition caused by activated MMPs. t-PA = Tissue plasminogen activator. Further information is provided in the text (graphic by M. Florczak-Rzepka and A. Sułkowska).
BBB destruction by MMPs during the inflammatory process based on Abbott et al. [37]. The process includes fibrinolytic cascade, oxidative stress, neutrophil infiltration and activation of immune cells, e.g. microglia. All involved cells along with stimulated endothelium release cytokines and chemokines as well as MMPs. Invasion into the brain is followed by destruction of TJ proteins and BM decomposition caused by activated MMPs. t-PA = Tissue plasminogen activator. Further information is provided in the text (graphic by M. Florczak-Rzepka and A. Sułkowska).
For a better understanding of the association between some MMPs and BBB breakdown, some facts need to be recalled. The remodeling of ECM in the whole organism by MMPs is guaranteed by the affinity of each group of MMPs to particular substrates. Interestingly, the ability to digest type IV collagen makes gelatinases A and B (MMP-2 and MMP-9, respectively) unique among other proteases and determines their role in BBB damage. In this case, binding to the BM is possible due to a fibronectin-binding region, a unique additional domain in the structure of this specific group of MMPs (fig. 1) [37]. Apart from this extracellular MMP function, there is also another, intracellular part.
Preservation of BBB function is guaranteed by tight junction (TJ) protein complexes within the endothelium (fig. 5). A TJ is a type of connection between two endothelial cells which limits transfer of substances and water through the BBB. The intercellular part of the TJ consists of claudins and occludins, which are attached to the intracellular scaffold made up of zona occludens proteins (1, 2, 3) and cingulin anchored in the cytoskeleton. As described by Cauwe and Opdenakker [15], MMPs were shown to attack zona occludens-1, causing its cleavage and TJ disruption.
Moreover, as already mentioned, MMPs may directly contribute to cellular death either by anoikis or by their intranuclear function. It was found that furin may activate MMPs inside cell nuclei and active enzymes are able to cleave PARP and PARP-activated X-ray repair cross-complementing protein-1 – both molecules take part in the DNA reparation process following oxidative stress damage [15].
Macrophage metalloelastase (MMP-12) has been found to be highly overexpressed in the area around hematoma. It was also demonstrated to be a key molecule that correlates with outcome in experimental ICH. Nevertheless, MMP-12 has been widely neglected in recent inquires and not yet considered in clinical investigations [38].
Interestingly, Mun-Bryce et al. [39] used somatic evoked potentials in hemorrhagic stroke to show the depressed cortical response, also contralateral to the hematoma site, which was followed by overexpression of MMP-9 and MMP-2 (gelatinases), implying that neuroinflammation triggered by blood extravasation in the course of ICH is not limited to the directly damaged brain tissue. This may suggest that the inflammation in the course of ICH is not only localized to the lesion but also involves distant areas.
Moreover, it has been postulated that the inflammatory cascade in hemorrhagic brain extends beyond it and is reflected in increased concentrations of inflammatory biomarkers in the peripheral blood. This is probably due to increased expression of cytokines such as TNF-α, IL-1β and specifically IL-6, which triggers production of acute-phase proteins in the liver such as C-reactive protein (CRP) and fibrinogen, thus amplifying inflammation (fig. 3) [40].
Summing up, MMPs play a crucial role in secondary brain injury following neuroinflammation, leading to opening of the BBB, apoptosis and both vasogenic and cytotoxic brain edema following ICH.
MMPs and Human Spontaneous ICH
Rosenberg [41] was one of the first to describe the causative connection between MMPs and stroke in rodents. Power et al. [38] described the activation profile and pathogenic aspects of MMP-2, -3, -7, -9 and -12 in the course of experimental ICH (table 1).
An overview of the function of MMPs derived from human spontaneous ICH studies and ICH experimental models

Clinically, MMPs have been associated with ischemic and hemorrhagic stroke. The MMP-9 concentration was found to predict hemorrhagic transformation after cerebral ischemia. The ability of MMP-9 to cleave the components of the BBB was proposed as a possible explanation for this correlation [42].
To the best of our knowledge, Abilleira et al. [43] were the first to describe overexpression of MMP-9 in human spontaneous ICH. They established the standard values for the plasma concentration of MMP-9 and other related biomarkers in healthy controls. Subsequently, they showed a direct correlation between MMP-9 plasma level, neutrophil count and relative perifocal edema (relative PHE = PHE/baseline spontaneous ICH) in deep hematomas, as well as neurological worsening measured by means of the Canadian Stroke Scale, all measured within 24 h after spontaneous ICH onset. This provides an important indication as to the significance of the BBB disruption of inflammatory origin in the course of spontaneous ICH. However, unlike in the case of deep hemorrhage, no significant relationship between the MMP-9 concentration and the course of lobar bleeding was found, probably due to the small sample size.
Early growth of the intracerebral bleeding in spontaneous ICH patients 48 h after onset was also proved to be associated with higher levels of MMP-9 and such molecular factors as TNF-α, IL-6 and fibronectin, as well as the common inflammatory markers leukocytes, platelets and fibrinogen [44].
Importantly, MMP-9 and MMP-2 were also detected in the perihematomal area in humans within 6 h after death, and their concentration was higher in the affected than in the contralateral hemisphere [36,45]. Wu et al. [46] confirmed the upregulation of MMP-9 in spontaneous ICH up to 5 days after hemorrhage onset. Additionally, NF-ĸB, p65 and macrophage inflammatory protein-2 were overexpressed, showing predominance at the hematoma site.
Another group carried out a prospective controlled study in 60 patients with spontaneous ICH who underwent hematoma evacuation [47]. The MMP-9 level was determined on days 1, 4 and 7 after neurosurgery in blood and cerebrospinal fluid (CSF). The MMP-9 level was higher in patients when compared with healthy controls in both blood and CSF. Interestingly, there was no MMP-9 found in normal CSF. Subsequently, these findings correlated positively with ICH and PHE volume as well as with the National Institutes of Health Stroke Scale score in the studied group of patients.
Moreover, there is a certain pattern of release of MMP-2, -3 and -9 after spontaneous ICH and thus also their inhibitors TIMP-1 and -2. The blood concentration of some of these molecules measured at different time points correlated with PHE volume and mortality within 3 months after spontaneous ICH onset [48]. Castellazzi et al. [49] found increasing MMP-9 serum levels in spontaneous ICH patients within a week from admission. At the same time, MMP-2 decreased continuously. Nevertheless, MMP levels correlated with PHE volume, which in turn increased during the observational period [9]. Hence, MMPs are likely to be involved in PHE formation and may have an impact on patients’ outcome. Thus, the time profile of MMP expression may be pivotal when thinking about future therapeutic aspects.
Yamani et al. [50] showed a relation between spontaneous ICH, enhanced MMP expression and subsequent development of coronary vasculopathy in patients who underwent heart transplant from spontaneous ICH donors. Increased MMP-2 and MMP-9 mRNA expression was found in the donors’ leukocytes and then in heart biopsies of the recipients 1 week after the surgical procedure. The MMP levels as well as the occurrence of coronary vasculopathy and myocardial fibrosis were significantly higher in patients whose donors died of ICH than in patients whose donors died due to trauma. These results provide further evidence supporting the important role of neuroinflammation after spontaneous ICH as a generalized process which may influence the outcome after ICH not only resulting from the secondary brain injury.
Wu et al. [46] investigated the timing of secondary brain damage in patients with spontaneous ICH who underwent hematoma evacuation. Apart from increased levels of MMP-9, they observed overexpression of caspase-3 and proliferation of terminal uridine nick-end labeling-positive cells as early as 6 h after spontaneous ICH onset with the tendency to progression up to 24 h, limited by the duration of the observation. This was accompanied by aggravated cell death and PHE, suggesting that secondary brain tissues damage occurs in the first few hours, at least up to 24 h after intracerebral bleeding onset and is primarily enhanced by apoptosis. Thus, the authors raised again the controversial problem of the relevance of early hematoma evacuation to improve patient outcome.
Spontaneous ICH is mainly associated with arterial hypertension. In a recent population study, arterial hypertension was found in 72.9% of patients with lobar ICH compared with 83.3% of cases presenting deep hemorrhages [1]. However, often in elderly spontaneous ICH patients with no history of arterial hypertension, cerebral amyloid angiopathy (CAA) is the most likely cause of the bleeding. Interestingly, it was found that amyloid, which leads to cerebral vessel pathology in CAA, is mainly composed of β-amyloid (the same as in Alzheimer’s disease) and can independently induce MMP-9, thus increasing the risk of spontaneous ICH in the elderly [51]. Hernandez-Guillamon et al. [52] studied MMP-2 and MMP-9 expression in patients with CAA-related hemorrhagic stroke and in healthy controls. Although there was no difference in MMP plasma levels between ICH patients and controls, MMPs were upregulated in the perihematomal area of brains in hemorrhagic patients when compared to the contralateral site of these brains and nonhemorrhagic brains. The authors also showed increased MMP-2 expression in β-amyloid-damaged vessels, whereas MMP-9 was found in inflammatory cells.
An important role of MMPs was also shown in the case of moyamoya disease. Fujimura et al. [53] found increased MMP-9 serum levels in patients diagnosed with this condition compared to normal MMP-9 levels found in healthy controls. This finding was not true for MMP-2, which remained at the same level in both groups. The authors concluded that this may facilitate pathological vascular processes and thus cause brain hemorrhages.
Finally, a deleterious role of MMPs was described by many authors in the case of subarachnoid bleeding (subarachnoid hemorrhage, SAH). Since SAH is mainly caused by aneurysm rupture, it is not considered spontaneous ICH and thus formally exceeds the scope of this review. Nevertheless, some most recent facts regarding the involvement of MMPs seem worth remarking upon. Frösen et al. [54] pointed out the role of inflammation and oxidative stress in the pathogenesis of intracranial aneurysm formation as the main cause of mural cell degeneration and impairment of endothelial cell function. Interestingly, the International Study of Unruptured Aneurysms group recently published a report on a possible protective effect of aspirin in unruptured aneurysms. In a group of 271 patients, they showed a trend toward a lower risk of hemorrhage due to aneurysm rupture in patients under continuous aspirin therapy (3 times weekly to daily) [55]. This supports a role of inflammation in the pathogenesis of subarachnoid bleeding. Chau et al. [56] studied CSF and blood concentrations of MMP-9 and white blood cell (WBC) counts in 35 SAH patients over 14 days. It was found that WBC and neutrophil count elevation over 2 weeks from SAH onset were independently associated with vasospasm. Also, WBC count and MMP-9 levels in the blood as well as CSF were related to poor clinical outcome (modified Rankin Scale score >2) within 3 months. They also found an association between WBC count and MMP-9 levels in the blood as well as between blood and CSF MMP-9 concentrations. This in turn supports the role of leukocyte recruitment but also MMP-9 activation as crucial for the neuroinflammatory cascade. Another group found a significant relationship between SAH severity as assessed by means of the World Federation of Neurosurgical Societies scale and pro-MMP-9 concentration in CSF. Also, the MMP-9 preform was remarkably higher in patients who developed cerebral ischemia as a complication of SAH [57].
Summing up, the role of MMPs in spontaneous ICH is evident. The early phase of the disease is characterized by a rapidly progressing inflammatory cascade, which leads to disruption of the BBB and spreads beyond the hematoma. The enlargement of bleeding along with the formation of brain edema determines the extent of the secondary brain damage (table 1). Moreover, it seems that the effect of the MMP-mediated response in the course of ICH may vary according to the MMP type. Prior studies imply that MMP-9 is involved in the acute phase, whereas MMP-2 acts more in the chronic phase, which is compatible with results of basic science studies. Moreover, neuroinflammation and thus MMPs play an important role in the pathogenesis of subarachnoid bleeding and its complications.
These data are supported by findings derived from animal models of spontaneous ICH highlighting the local origin of the inflammatory cascade, which disperses afterwards. In all experimental studies presented in table 1, either collagenase-induced ICH models or models with autologous blood injection into rodent brains were used.
MMPs as Targets for Therapy
Given the unquestionable role of MMPs and foremost the gelatinases in the pathology of spontaneous ICH, their inhibition becomes an interesting issue regarding therapy of spontaneous ICH. So far, MMPs attenuation has been studied in a few experimental models of spontaneous ICH (table 2). Interestingly, most of the inhibitors tested are derived from cancer research and were proved to act as possible antimetastatic agents. However, they are mostly unspecific, which may explain their broad spectrum of function including some inevitable side effects mainly related to long-term suppression of the essential physiologic processes mediated by MMPs but also to inhibition of ADAMs and ADAMs with thrombospondin motifs [21]. The most important MMPIs along with their brief profile in relation to ICH are listed in table 2.

Wang et al. [58] tested GM6001, a broad-spectrum metalloproteinase inhibitor administered intraperitoneally (100 mg/kg) in mice every 24 h for 2 days starting 2 h after ICH induction. They managed to demonstrate a significant reduction of the secondary brain damage due to leukocyte infiltration, MMP-9 activity and oxygen-derived free radicals (reactive oxygen species). As a result, brain edema, neuronal loss and neurological deficit diminished on day 3 after ICH onset and treatment application, confirming the crucial role of MMP-9 in the pathology of the early deterioration after ICH.
Lee et al. [59] showed a link between MMP-9 but not MMP-2 overexpression in mouse brain tissues and vascular endothelial growth factor-induced brain hemorrhage. In this model imitating brain arteriovenous malformation-derived hemorrhage, minocycline suppressed ICH evolution, including significant reduction of the bleeding. A slightly smaller effect on hematoma was observed when using pyrrolidine dithiocarbamate, inhibitor of NF-ĸB, an upstream regulator of MMPs. These findings confirm that MMP-9 expression due to ICH can be regulated through different pathways, which is even more challenging in terms of possible inhibitory therapy.
Quite interesting findings derive from another experimental study where the authors tested the following nonspecific and specific MMPIs: BB-1101, BB-94, BB-2983, IW499, tetracyclines, steroids and DMSO (a selective MMP-2 and MMP-9 inhibitor). A significant reduction in BBB permeability was achieved when using BB-1101, BB-94, minocycline and 10% DMSO. Other substances, including GM6001 and the sole MMP-2 inhibitor IW499, were ineffective and did not reduce the leakage through the BBB [41].
On the other hand, Grossetete et al. [60] showed BB-94 to increase hematoma with no effect on PHE. More importantly, it also enhanced cell death.
Although so far not considered in the group of MMPIs, there are some agents that may be worth noting, as described below.
Memantine, an uncompetitive N-methyl-d-aspartate receptor antagonist, reduced neuroinflammation, including MMP-9 levels, and caused hematoma reduction by 47% [61].
Sinn et al. [62] demonstrated valproic acid to enhance alleviation of the inflammatory cascade and significantly reduce bleeding as well as attenuate cell death in the perihematomal area after ICH in rats. The rationale provided refers to the ability of valproic acid to regulate gene expression and thus the possible alteration of the cytokine profile involved in the pathophysiology of ICH.
Final Considerations and Conclusions
Many data indicate the deleterious role of MMPs in the secondary brain damage following spontaneous ICH. At present, available clinical studies are scarce, mostly based on small populations and thus deliver merely a limited insight into the topic. However, the important role of MMPs in human spontaneous ICH in regard to the clinical and radiological course of the disease as well as its impact on functional outcome seems to be already established. A great number of rodent ICH studies strengthen this thesis by delivering promising data on MMPIs and MMP-9 knockout mice [45]. Still, there are some apparently inconsistent results from other knockout experiments suggesting that the total blockade of MMPs may not be unquestionably advantageous [63,64]. However, some of these data are based on different experimental models of ICH, and thus conclusions should be drawn very carefully. Another reason for contrary findings may be the above-mentioned low specificity of many drugs tested for MMP inhibition.
It should not be forgotten that MMPs as natural molecules in the human body are involved in a number of processes, which comprise other vital functions apart from the inflammatory cascade [14,24,27,65].
Hence, when considering blocking MMPs for therapeutic purposes, one has to be aware of their ubiquitous role in the human body and weigh the pros and cons carefully. The activity of MMPs, though, as crucial for the recovery phase after ICH, cannot be irreversibly blocked but merely modulated. Therefore, more properly designed experimental studies are needed to define the critical point of the MMP-activating cascade before any conclusions can be transferred to clinical trials.
Acknowledgements
We wish to express our special thanks to Agnieszka and Bartosz Sułkowscy for their professional help in visualizing the heart of the matter.
