

Abstract
We characterize a novel otoferlin mutation discovered in a sibling pair diagnosed with auditory neuropathy spectrum disorder and investigate auditory nerve function through their cochlear implants. Genetic sequencing revealed a homozygous mutation at the otoferlin splice donor site of exon 28 (IVS28 + 1G>T) in both siblings. Functional investigation showed that the intronic sequence between exons 28 and 29 was retained in the mutated minigenes that were expressed in 293T cells. Auditory nerve compound action potential recovery functions in the siblings demonstrated different rates of neural recovery, with sibling AN1 showing rapid recovery (1.14 ms) and AN2 showing average recovery (0.78 ms) compared to subjects with sensorineural hearing loss (average: adults 0.71 ms, children 0.85 ms). Differences in neural recovery were consistent with speech perception differences between the siblings. Genotype information may indicate site of lesion in hearing loss; however, additional, as yet, unknown factors may impact clinical outcomes and must be considered.
Introduction
Auditory neuropathy spectrum disorder (ANSD) is a hearing disorder characterized by present outer hair cell function and dys-synchronous neural activity [Starr et al., 1996]. From this relatively broad clinical definition, ANSD encompasses several potential etiologies including pre- and postsynaptic sites of lesion. Determining site of lesion in ANSD has the potential to guide clinical intervention to optimize patient outcomes, and genetic testing may contribute to this purpose. One candidate gene is otoferlin (OTOF), as associations between mutations in OTOF and ANSD have been established [Chiu et al., 2010; Rodriguez-Ballesteros et al., 2003, 2008; Varga et al., 2003, 2006]. OTOF is a protein expressed in cochlear inner hair cells, and genetic mutations resulting in anomalous protein can give rise to clinical findings of ANSD [Rodriguez-Ballesteros et al., 2003, 2008; Varga et al., 2003, 2006]. In addition, OTOF expression in inner hair cells and its critical role in vesicle exocytosis have been characterized [Roux et al., 2006; Yasunaga et al., 1999, 2000]. Therefore, in cases of ANSD with an OTOF mutation, the site of lesion is assumed to be presynaptic and auditory nerve function is presumed to be intact [Loundon et al., 2005; Rouillon et al., 2006; Roux et al., 2006; Santarelli et al., 2009].
Although genetic testing may indicate site of lesion, investigations of the relationships between genotype and phenotype are critical for clinical use of genetic information. One question is the integrity of the neural system in patients with ANSD, which can be measured using electrical stimulation through a cochlear implant. Stimulation through a cochlear implant elicits the electrically evoked auditory brainstem response and electrically evoked compound action potential (ECAP) in patients with ANSD [Buss et al., 2002; Fulmer et al., 2011; Kim et al., 2011; Mason et al., 2003; Peterson et al., 2003; Runge-Samuelson et al., 2008; Shallop et al., 2001, 2004]. ECAPs are commonly used in the clinical setting to objectively measure neural threshold and response growth to different levels of electrical stimuli. In addition, ECAPs can provide information regarding the temporal response properties of the auditory nerve [Brown et al., 1990]. This is of particular interest in ANSD, as dys-synchronous neural responses to acoustic stimulation underlie the temporal processing issues that characterize this disorder [Rance et al., 2004; Zeng et al., 1999, 2005]. By measuring auditory nerve responses to pulses presented at different rates, ECAP recovery functions indicate how quickly the auditory nerve recovers from electrical stimulation. Previous studies have found that children with ANSD have a similar rate of ECAP recovery as children with sensorineural hearing loss (SNHL) [Fulmer et al., 2011; Kim et al., 2011]. However, site of lesion (i.e. pre- or postsynaptic) for children in these studies was not known, and therefore it was not possible to determine whether this was a relevant factor.
The purpose of this study was to describe a novel OTOF splice-site mutation identified in a sibling pair diagnosed with ANSD, and investigate rate of neural recovery to electrical stimulation in these siblings.
Methods
Subjects
All subjects gave informed consent for their participation in the study. The study protocol was approved by the Human Research Review Committee of the Children's Hospital of Wisconsin/Medical College of Wisconsin. Two Caucasian female siblings clinically diagnosed with ANSD participated in the genetic and phenotypic testing for this study. Both siblings were implanted unilaterally with Advanced Bionics HiRes 90k devices; AN1 received the Helix electrode array and AN2 received the HiFocus 1J electrode array. To compare the siblings' ECAP recovery functions to those with SNHL, data from 3 pediatric (n = 5 ears) and 12 adult (n = 12 ears) subjects with SNHL who were also implanted with Advanced Bionics CII or HiRes 90k devices were included. Electrode array types for the control subjects included HiFocus 1J (n = 8), Helix (n = 2), HiFocus with Positioner (n = 3), and HiFocus without Positioner (n = 4). Recovery function and speech perception data for the siblings were included in ANSD group data previously reported by our lab [Fulmer et al., 2011].
Mutation Detection
Blood was obtained from AN2 during cochlear implant surgery. DNA was isolated from blood using the Puregene DNA Isolation Kit (Life Technologies, Grand Island, N.Y., USA) and sent to the Molecular Otolaryngology Research Laboratory at the University of Iowa for sequencing of the 48 OTOF exons and adjacent intronic sequences. Subsequent single nucleotide polymorphism (SNP) analysis for AN1 was performed using DNA obtained from buccal swabs and isolated using the Puregene Blood Core Kit (Life Technologies, Grand Island, N.Y., USA).
Minigene Construction, Transfection, and RNA Analysis
After a novel mutation was identified in the 5′ splice site (ss) of OTOF exon 28 in the siblings, a minigene of the region surrounding the mutation was constructed to examine the potential functional implications on RNA splicing. The OTOF minigene consisted of exons 27 through 30 and the beginning of intron 30, and was generated by PCR from wild-type genomic DNA using primers otoEco-f (aagcttgcggccgcgaattct) and otoSma-r (cccggggcagatagtctggttca). The resulting 1,664 bp product was cloned into the Eco RI-Xma I sites of the p3XFLAG-CMV-7.1 expression vector (Sigma, St. Louis, Mo., USA) to generate p3XFLAG-oto1. A version of the minigene was created that precisely ended with exon 30 (p3XFLAG-oto2) by digesting the plasmid with Kpn I-Xma I and inserting an oligonucleotide with the exon 30 end sequence. A mutant version of the minigene representing that observed in the siblings was made by introducing an exon 28 5′ ss G to T mutation by overlap PCR. An upstream fragment was generated with primers Oto198-for (caccaggagccagcagcagac) and oto5′mut-rev (cctgcactcaacacttcaaacacttgacg), and a downstream fragment with oto1056-rev (ctctggtcgcggcttggactg) and oto5′mut-for (cgtcaagtggtttgaagtgttgagtgcagg), and the fragments combined in an overlap PCR reaction to produce a mutant fragment. This fragment was digested with PasI and the 780-bp fragment was used to replace the wild-type Pas I fragment. The mutant sequence was verified by DNA sequencing. The minigenes were transfected into 293T and HeLa cells using the calcium phosphate method, and 2 days after transfection, RNA was isolated using RNAeasy columns (Qiagen) and DNase I treated. For RT-PCR, 1 µg of RNA was reverse transcribed in a 20-µl reaction using oligo dT as a primer, and for PCR, 1.0 µl of RT mix was subjected to PCR with 1R and Sma REV for 25 cycles at 95°C for 45 s, 54 for 45 s, and 72 for 45 s. Vector DNA served as a positive control (which also served as a marker for unspliced RNA) and water was a negative control. PCR products were resolved on a 1% agarose gel and stained with EtBr. The bands labeled 1 and 2 in figure 3 were excised from the gel and sequenced to determine the nature of the PCR products.
ECAP Recovery Functions
ECAP recovery functions were obtained using the methods previously employed in our lab [Fulmer et al., 2011; Lee et al., 2012]. In summary, stimuli were delivered to the subjects using the Bionic Ear Data Collection System (BEDCS, software version 1.16.191) provided by Advanced Bionics Corporation (Sylmar, Calif., USA) through a research-dedicated clinical Platinum Series Processor and Clarion Programming Interface. Stimuli were two monopolar biphasic pulses of 21.6 µs per phase delivered to electrode 8 and recorded from electrode 6. The two pulses, i.e. masker and probe, were separated by increasing lengths of time, or interstimulus intervals (ISIs), ranging from 0.2 to 7.0 ms. The masker was presented at maximum comfort level (MCL) and the probe at 80% MCL. Amplitudes of ECAP responses to the second pulse were plotted as a function of ISI and normalized to the ECAP amplitude at 7.0 ms ISI [Brown et al., 1996]. The recovery functions were curve-fitted with an exponential growth function, and the exponent value determined the recovery constant for each subject. Higher recovery constant values indicated faster ECAP recovery. ECAP recovery functions were collected when the siblings were 5 (AN1) and 4 (AN2) years of age.
Speech Perception Testing
Speech perception scores from routine clinical testing at the 3-year postimplantation interval were obtained from the siblings' patient records. Spoken word perception was measured using the Lexical Neighborhood Test Easy [Kirk et al., 1995] administered with live voice in a quiet condition.
Research speech recognition thresholds (SRT) in quiet and noise were obtained during the same session as the ECAP recovery testing. SRTs were measured using the Children's Realistic Index for Speech Perception Junior [Garadat and Litovsky, 2007]. The Children's Realistic Index for Speech Perception Junior is a closed-set, 4-alternative forced-choice task that consists of 16 monosyllable and spondee words likely to be in the vocabulary of an average 2.5- to 3-year-old child. Starting with a word presented at 60 dB SPL, the level was changed adaptively to determine SRT, defined as 79.4% correct on the psychometric function [Levitt, 1971]. In the noise condition, the competing stimuli were 2-talker babble sentences presented at a constant overall level of 45 dB SPL. Smaller SRT values indicate better speech perception performance.
Results
Case Descriptions
AN1 was born full-term by emergency C-section subsequent to maternal infection/fever and high fetal heart rate. She was in the NICU for 1 week after delivery due to infection and respiratory distress, and was found positive for cystic fibrosis. AN1 was identified with hearing loss at 2 weeks of age, and follow-up objective audiologic tests performed at 4 weeks and at 4 months of age revealed absent auditory brainstem response with present cochlear microphonic to click stimuli presented at 80 and 102 dB nHL, and present distortion product otoacoustic emissions (DPOAEs) in both ears, resulting in a diagnosis of bilateral ANSD. Behavioral audiometric testing revealed bilateral profound hearing loss (fig. 1a). Hearing aids were fitted at 4 months of age, and although she wore the hearing aids consistently (i.e. 10 h/day), aided benefit was reportedly limited to inconsistent increase in sound awareness. The profound hearing loss combined with minimal hearing aid benefit identified her as a cochlear implant candidate. She received a cochlear implant in her right ear at age 18 months.

Preoperative audiograms for siblings AN1 (a) and AN2 (b). Hearing level in dB is plotted as a function of frequency in Hz. RE refers to the right ear (circle symbols), and LE refers to the left ear (X symbols). A symbol with a downward arrow indicates no response at that frequency.
Preoperative audiograms for siblings AN1 (a) and AN2 (b). Hearing level in dB is plotted as a function of frequency in Hz. RE refers to the right ear (circle symbols), and LE refers to the left ear (X symbols). A symbol with a downward arrow indicates no response at that frequency.
AN2 was born at full-term with no significant birth history. Clinical records indicated that newborn hearing screening was performed at another facility using only OAEs, which AN2 passed. Her mother suspected hearing loss at 9 months of age and brought her to our facility for assessment. Comprehensive objective testing showed absent auditory brain response with present cochlear microphonic to click stimuli presented at 80 and 90 dB nHL. Audiometric thresholds were in the severe-to-profound hearing loss range bilaterally (fig. 1b). Hearing aids were fitted at 11 months of age and AN2 demonstrated limited benefit. She received a cochlear implant in her right ear at 16 months of age.
Splice-Site Mutation Identification
To determine the basis of the patient phenotypes, DNA sequencing was performed on genes associated with ANSD. Both siblings were negative for GJB2 mutations. Analysis of DNA from AN2 revealed a homozygous mutation at the splice donor site for exon 28 (IVS28 + 1G>T) (fig. 2) of OTOF. Follow-up SNP analysis for AN1 in our laboratory confirmed homozygosity for the mutation in this sibling. A recent search showed that this mutation was not part of the SNP database (http://www.ncbi.nlm.nih.gov/SNP/; searched 1/9/2013).
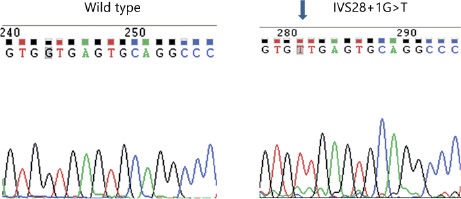
DNA sequencing electropherograms of the OTOF exon 28 5′ ss region from a normal individual (wild-type, left) and AN2 (right). The G to T mutation site is indicated by the arrow. The normal G and mutant T at this location is highlighted in grey in both panels.
DNA sequencing electropherograms of the OTOF exon 28 5′ ss region from a normal individual (wild-type, left) and AN2 (right). The G to T mutation site is indicated by the arrow. The normal G and mutant T at this location is highlighted in grey in both panels.
Minigene Analysis
The IVS28 + 1G>T mutation suggested that altered mRNA splicing of OTOF mRNA might underlie the siblings' ANSD phenotype. Because patient tissue samples could not be analyzed directly, the effect of the mutation on OTOF splicing was assessed in tissue culture cells using an OTOF minigene. Minigenes containing exon 28-30 and intronic sequences were amplified from wild-type genomic DNA and cloned into a mammalian expression vector. The patient mutation was introduced by site-directed mutagenesis (fig. 3a). Analysis of RNA from transfected 293T cells using primers specific for minigene mRNA showed two bands from the wild-type construct that, upon excision and sequencing, corresponded to completely spliced mRNA (upper band) and a lower band that represents RNA that used a cryptic 3′ss in exon 30 that has not been reported in humans and, thus, is likely to be an artifact of the minigene system (fig. 3b). For the mutant, the bands corresponding to fully spliced species were lost and replaced by bands that, upon sequencing, corresponded to retention of intron 28. Retention of intron 28 did not change the reading frame of mutant OTOF mRNA, and resulted in 37 additional amino acids. Similar results were obtained upon transfecting HeLa cells (data not shown). These results are consistent with a splicing defect caused by IVS28 + 1G>T.
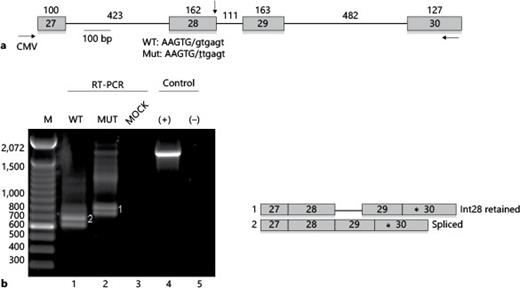
IVS28 + 1G>T causes aberrant splicing of an OTOF minigene. a Schematic representation of the OTOF minigene. Boxes represent exons, and the lines represent introns. The sizes of the exons and introns are indicated. The horizontal arrows indicate PCR primers for RT-PCR detection of minigene RNA: CMV is a primer to a vector-derived portion of the minigene RNA. The 5′ splice site of exon 28 is indicated with a vertical arrow, and shown below is the sequence of the wild-type and mutant 5′ splice sites, with exon sequences in capital letters, the junction denoted by the slash, and intron sequences in lower case. The mutation is underlined. b RT-PCR analysis of RNA isolated from 293T cells transfected with the wild-type (WT) and mutant (mut) minigenes. Mock, mock-transfected cells. Controls were the wild-type plasmid DNA (+) and water was the negative control (-). To the right are depictions of the RNA forms represented by the PCR products. The asterisks denote a cryptic 3′ss that generates the lower band in each lane, which was determined by DNA sequencing of the excised bands.
IVS28 + 1G>T causes aberrant splicing of an OTOF minigene. a Schematic representation of the OTOF minigene. Boxes represent exons, and the lines represent introns. The sizes of the exons and introns are indicated. The horizontal arrows indicate PCR primers for RT-PCR detection of minigene RNA: CMV is a primer to a vector-derived portion of the minigene RNA. The 5′ splice site of exon 28 is indicated with a vertical arrow, and shown below is the sequence of the wild-type and mutant 5′ splice sites, with exon sequences in capital letters, the junction denoted by the slash, and intron sequences in lower case. The mutation is underlined. b RT-PCR analysis of RNA isolated from 293T cells transfected with the wild-type (WT) and mutant (mut) minigenes. Mock, mock-transfected cells. Controls were the wild-type plasmid DNA (+) and water was the negative control (-). To the right are depictions of the RNA forms represented by the PCR products. The asterisks denote a cryptic 3′ss that generates the lower band in each lane, which was determined by DNA sequencing of the excised bands.
ECAP Recovery Functions
Figure 4 shows the ECAP recovery waveforms and functions for AN1 (top) and AN2 (bottom). Although the stimuli were presented at MCL for both subjects, ECAP waveforms for AN1 we of larger absolute amplitude compared to AN2. Rate of neural recovery was quantified from the ECAP recovery functions for all subjects, and recovery constants are shown for the respective groups (fig. 5). Average recovery constants for the adult and pediatric groups with SNHL were 0.71 ms (SD 0.20) and 0.85 ms (SD 0.32), respectively. There was no significant difference between the SNHL control groups (p = 0.29). AN1 had a recovery exponent of 1.14 ms, which was 1 SD higher than the average of the pediatric subjects, and more than 1 SD higher than the average of the adult subjects. AN2 had a recovery exponent of 0.78 ms, which was within the average range of both SNHL groups. A one-way ANOVA was used to test for effects of electrode array type on ECAP recovery constants, and no significant effects were found (F(3, 15) = 0.76, p = 0.53).

ECAP waveforms (left) and recovery functions (right) are shown for subjects AN1 (top) and AN2 (bottom). Waveform amplitudes are indicated with scale bars.
ECAP waveforms (left) and recovery functions (right) are shown for subjects AN1 (top) and AN2 (bottom). Waveform amplitudes are indicated with scale bars.

Recovery exponents for subjects with SNHL (adults, circles; children, squares) and siblings with ANSD. Average exponent values for the SNHL groups are indicated by stars.
Recovery exponents for subjects with SNHL (adults, circles; children, squares) and siblings with ANSD. Average exponent values for the SNHL groups are indicated by stars.
Speech Perception
The Lexical Neighborhood Test Easy spoken word scores obtained clinically at the same 3-year postimplantation interval were 80% correct for AN1 and 44% correct for AN2. This indicated better speech perception performance with the same length of implant use for AN1 compared to AN2.
The SRT results in both quiet and noise conditions were consistent with the differences observed in clinical spoken word perception scores. In quiet, SRTs were 44 dB (AN1) and 65 dB (AN2); SRTs in noise were 52 dB (AN1), and 70 dB (AN2).
Discussion
OTOF Splice-Site Mutation in AN1 and AN2
We have identified a novel splice-donor site mutation in the OTOF gene that is the likely cause of ANSD in this sibling pair. An effect of IVS28 + 1G>T on OTOF mRNA splicing was demonstrated using a minigene system. Mutations in 5′ splice sites are associated with many human diseases and conditions [Cooper et al., 2009] and can result in exon skipping, activation of cryptic 5′ splice sites, or skipping of the exon, with exon skipping predominating [Nakai and Sakamoto, 1994]. Exons are typically small (less than 300 nt) with an average of ∼170 nt, whereas introns are more variable in length, with an average of ∼5,400 nt [Sakharkar et al., 2004]. The unit of recognition by the splicing apparatus is typically the exon via ‘exon definition' [Berget, 1995], which predicts that 5′ ss mutations will result in exon skipping. However, the size of flanking introns also influences splicing, and small introns can undergo ‘intron definition' [Hertel, 2008; Sterner et al., 1996] in which a 5′ ss mutation might result in intron retention. Consistent with this, analysis of the mutant OTOF minigene expression showed that intron 28 retention was the predominant outcome (fig. 3). Therefore, the additional 37 amino acids contributed by retained intron 28 are predicted to compromise protein function.
Audiologic Phenotype
Audiologic phenotypes for most known OTOF mutations show severe to profound hearing loss [Adato et al., 2000; Choi et al., 2009; Houseman et al., 2001; Migliosi et al., 2002; Mirghomizadeh et al., 2002; Rodriguez-Ballesteros et al., 2003, 2008; Romanos et al., 2009; Rouillon et al., 2006; Tekin et al., 2005; Varga et al., 2003, 2006; Yasunaga et al., 1999, 2000], although some OTOF mutations are associated with moderate to severe hearing loss [Chiu et al., 2010; Varga et al., 2003, 2006] or fluctuating hearing levels with changes in temperature [Marlin et al., 2010; Starr et al., 1998; Varga et al., 2006]. At this time, all reports of splice-site OTOF mutations indicate a severe to profound hearing loss phenotype [Adato et al., 2000; Rodriguez-Ballesteros et al., 2008; Rouillon et al., 2006; Varga et al., 2003, 2006], which is consistent with the audiologic findings for siblings AN1 and AN2.
ECAP Recovery Functions and Speech Perception
Comparisons of electrically evoked neural potentials with cochlear implant stimulation show similar responses between subjects with ANSD and SNHL, including presence of electrically evoked auditory brainstem response and ECAP and similar slopes of ECAP recovery functions [Fulmer et al., 2011; Kim et al., 2011]. However, there is evidence for variability in electrically evoked neural response characteristics in the ANSD population. For example, while intraoperative electrically evoked auditory brainstem response may be present in children with both ANSD and SNHL, quantitative analysis of the waveforms has shown reduced wave V suprathreshold amplitudes in patients with ANSD [Runge-Samuelson et al., 2008], possibly indicating residual dys-synchronous neural activity central to the auditory nerve. In previous investigations of subjects with ANSD the site of lesion is often unknown, which may contribute to the variability in findings within this population. In the present study, the siblings shared the same genotype, and presumably the same presynaptic site of lesion. It could be hypothesized that the presynaptic lesion would give rise to neural recovery that would be similar between the siblings, and may differ from those with SNHL of various etiologies. Only sibling AN1 had recovery that was ≥1 SD faster than the average recovery of the SNHL groups, while the recovery rate for AN2 was within the average ranges of the SNHL controls. Therefore, despite having an identical OTOF mutation, there were differences in neural responses between siblings.
A potential difference may include the size of the stimulated neural population, as ECAP recovery rate has been reported to be influenced by this factor [Botros and Psarros, 2010b]. Electrode array type and stimulation level have also shown effects on ECAP recovery [Botros and Psarros, 2010b] and the relationship between ECAP thresholds and psychophysical loudness profiles [Botros and Psarros, 2010a]. Botros and Psarros [2010b] showed effects of electrode array type on ECAP recovery function characteristics between the nucleus contour and straight arrays, with the contour demonstrating significantly slower neural recovery with greater temporal responsiveness, and potentially greater neural recruitment, than the straight array. While the siblings were both tested at the same loudness level (MCL), they had different electrode array types (AN1 Helix and AN2 HiFocus 1J). The electrode array types for the Advanced Bionics devices in this study did not show a significant effect on ECAP recovery rate; however, the sample sizes for each array type were relatively small, and specific investigation into these relationships would be beneficial. The Advanced Bionics Helix array was designed for more modiolar placement, and the HiFocus 1J was designed for more lateral placement. In this respect, the ECAP recovery rates for the siblings were inconsistent with the findings with the nucleus contour (modiolar) and straight (lateral) arrays, with AN1 showing faster recovery with a Helix than AN2 with the HiFocus 1J. This is an anecdotal observation, however, given the small sample size.
Speech perception performance with a cochlear implant also differed between the siblings, with AN1 demonstrating better performance than AN2 across all conditions, including spoken word perception in quiet, and SRT in quiet and in noise. In the study by Fulmer et al. [2011], recovery rate was compared to SRT in quiet and in noise for children with ANSD and SNHL. There was no relationship between speech perception and recovery rate for the ANSD and SNHL groups when analyzed separately, nor for the groups together for speech perception in quiet. However, there was a significant relationship between speech perception in noise and recovery rate when all subjects were analyzed together. Relative to the ANSD group data in Fulmer et al. [2011], AN1 showed better than average SRT in quiet and represented best performance of the group in noise. SRTs for AN2 showed the poorest performance for both quiet and noise conditions. Therefore, for the siblings, rate of neural recovery was consistent with speech perception performance, although caution in interpretation is necessary given the small sample size.
Relationships between rate of neural recovery and speech perception have been observed in cochlear implant users [Brown et al., 1990; Kiefer et al., 2001], although some investigations have not seen this relationship [Abbas and Brown, 1991]. It is possible, however, that performing more comprehensive assessments beyond presence or absence of neural responses for all patients may help with intervention. There is evidence that adjusting CI programming parameters, such as lowering stimulation rate, has the potential to facilitate development of auditory skills for patients who struggle with speech perception abilities, particularly those with ANSD and/or comorbid developmental conditions [Pelosi et al., 2012; Peterson et al., 2003].
Conclusions
This study identified a novel OTOF splice-site mutation in two siblings with ANSD. While this study included a small number of subjects, there are general factors to consider when attempting to use genotype to predict phenotype. While genotype may indicate site of lesion, additional factors impact clinical outcomes. There is a need to consider alternative aspects even when the ‘causal' genotype is known, and in ANSD a pre-synaptic site of lesion does not necessarily rule out neurological and/or central pathologies. This study also indicates that performing additional testing beyond what is typically done clinically may provide additional phenotypic information for individual patients. The additional information may inform clinical intervention approaches and parameter settings to optimize outcomes.
Acknowledgments
This study was funded by NIH/NIDCD K23 DC008837 (Runge, PI). We would like to thank Jamie Jensen, AuD, and Sarah Klajbor, AuD, who provided valuable assistance in collecting clinical information and performing research data collection. Ruth Y. Litovsky, PhD, generously provided the Children's Realistic Index for Speech Perception Junior research software. Leo Litvak, PhD, provided the BEDCS programs for recording ECAP recovery functions. We appreciate the support of the Koss Cochlear Implant Program at the Medical College of Wisconsin, Milwaukee, Wisc., USA, and the Masters Family Speech and Hearing Center at Children's Hospital of Wisconsin, Milwaukee, Wisc., USA.
Disclosure Statement
Dr. Runge serves on the Audiology Advisory Boards for Advanced Bionics Corp. and MED-EL Corp. Dr. Friedland serves on the Surgical Advisory Board for MED-EL Corp.
References
This work was conducted at the Medical College of Wisconsin, Milwaukee, Wisc., USA.
