

Abstract
Background/Aims: Angiotensin converting enzyme 2 (ACE2) treatment suppresses the severity of acute lung injury (ALI), through antagonizing hydrolyzing angiotensin II (AngII) and the ALI-induced apoptosis of pulmonary endothelial cells. Nevertheless, the effects of ACE2 on vessel permeability and its relationship with vascular endothelial growth factor a (VEGFa) remain ill-defined. In the current study, we examined the relationship between ACE2 and VEGFa in ALI model in mice. Methods: Here, we used a previously published bleomycin method to induce ALI in mice, and treated the mice with ACE2. We analyzed the levels of VEGFa in these mice. The mouse lung vessel permeability was determined by a fluorescence pharmacokinetic assay following i.v. injection of 62.5µg/kg Visudyne. VEGFa pump or SU5416 pump was given to increase or decrease VEGFa effects, respectively. The long-term effects on lung function were determined by measurement of lung resistance using methacholine. Results: ACE2 treatment did not alter VEGFa levels in lung, but antagonized the effects of VEGFa on increases of lung vessel permeability. Ectogenic VEGFa abolished the antagonizing effects of ACE2 on the vessel permeability against VEGFa. On the other hand, suppression of VEGF signaling mimicked the effects of ACE2 on the vessel permeability against VEGFa. The suppression of vessel permeability resulted in improvement of lung function after ALI. Conclusion: ACE2 may antagonize the VEGFa-mediated increases in lung vessel permeability during ALI, resulting in improvement of lung function after ALI.
Introduction
Acute lung injury (ALI) is characterized with expiratory dyspnea, refractory hypoxemia and non-cardiogenic pulmonary edema, the deterioration of which could result in the aggravation of ALI into highly lethal Acute Respiratory Distress Syndrome (ARDS) [1,2,3,4,5,6,7,8,9,10]. The cellular pathology of ALI is comprised of loss of alveolar-capillary membrane integrity, excessive transepithelial neutrophil migration and release of pro-inflammatory cytokines, e.g. interleukin (IL)-6, tumor necrosis factor (TNF)-α and CXCL1 [1,2,3,4]. Injuries of both pulmonary endothelial cells (PECs) and alveolar epithelial cells occur after ALI, resulting in loss of respiratory capacity [1,2,3,4]. Among all these pathological processes, the increases in lung vessel permeability are critical and play a non-redundant role in the pathogenesis of ALI, which allows penetration of neutrophils cross the vascular epithelia to cause the pathological changes [1,2,3,4]. Excessive and prolonged activation of neutrophils results in basement membrane destruction and increased permeability of the alveolar-capillary barrier [1,2,3,4]. Moreover, neutrophils also release pro-inflammatory and pro-apoptotic cytokines to injure adjacent cells to create ulcerating lesions to aggravate the damages of the alveolar-capillary barrier [1,2,3,4].
The vessel permeability is mainly regulated by a coordination of a series of pro-angiogenic and anti-angiogenic factors, among which the most potent pro-angiogenic factor is vascular endothelial growth factor a (VEGFa) [11,12,13,14,15], which has also been shown to play a critical role in the increases in lung vessel permeability in ALI [16,17,18]. VEGFa exerts its function by binding to its main receptor, VEGF receptor 2 (VEGFR2). In renin-angiotensin system, renin induces the production of angiotensin I (Ang I), which is converted to an important vasoconstrictive peptide Ang II by Ang I-converting enzyme (ACE) [19]. Angiotensin-converting enzyme 2 (ACE2) is the homolog of ACE but counterbalances the ACE activity through induction of degradation of Ang II [19]. Recently, we and others have shown that ACE2 has therapeutic effects on ALI, seemingly through different mechanisms, e.g. suppression of apoptosis of PECs [20,21,22,23,24,25,26]. However, a role of ACE2 in antagonizing VEGFa-mediated increases in lung vessel permeability in ALI has not been acknowledged.
In the current study, we examined the relationship between ACE2 and VEGFa in ALI model in mice. We found that ACE2 treatment did not alter VEGFa levels in lung, but antagonized the effects of VEGFa on increases of lung vessel permeability. Ectogenic VEGFa abolished the antagonizing effects of ACE2 on the vessel permeability against VEGFa. On the other hand, suppression of VEGF signaling mimicked the effects of ACE2 on the vessel permeability against VEGFa. The suppression of vessel permeability resulted in improvement of lung function after ALI.
Materials and Methods
ALI model in mice and ACE2 treatment
All mouse experiment protocols were approved by the Animal Research and Care Committee at Shanghai Jiaotong University. All experiments were performed in accordance with the guidelines from the Animal Research and Care Committee at Shanghai Jiaotong University. Mouse manipulations were performed in accordance with the principles of laboratory care, supervised by a qualified veterinarian. The methods were carried out in accordance with the approved guidelines.
Specific pathogen free (SPF) Balb/c mice (aged 10 weeks, weight 18 - 22g) were supplied by Laboratory Animal Center of Shanghai Academy of Sciences, Chinese Academy of Sciences, China. After acclimatization for 1 week, a group of mice were used as the no-injury group and received no treatments (NT). The lung injury model was induced in another groups of mice, by intraperitoneal administration of the bleomycin solution (10mg/mL, Sigma-Aldrich, St Louis, MO, USA) at 20 mg/kg body weight, as has been applied and described before [20,21,22,23,24,25]. This ALI model is stable and easy to be built. No death was observed in the current study. After successful induction of ALI, the mice were further randomly divided into groups to receive single tail vein injection of either saline (termed ALI), or ACE2 (R&D systems, Shanghai, China) at a dose of 1mg/kg body weight (termed ALI+ACE2). Ten mice were used in one experimental group.
VEGFa or SU5416 pump treatment
To increases VEGFa levels or suppresses VEGFa, 10µg recombinant VEGFa (Sigma-Aldrich) or 2 µg SU5416 [27,28] (Sigma-Aldrich) was dissolved in PBS and placed in mini-osmotic pumps (Alzet osmotic pumps, Cupertino, CA, USA), which were implanted subcutaneously to the mice, at the time of ACE2 treatment. Mini-pumps containing PBS only were implanted in the mice (CTL pump) as a control. The pumps keep releasing solution for 7 days. The mice were analyzed after 7 days.
Assessment of vessel permeability
The mouse was anaesthetized and a left-sided thoracotomy was performed through the 4th intercostal space to allow the left lung mobilized under the microscope. A left cervical incision was performed to cannulate the external jugular vein. Visudyne (Novartis, Hettlingen, Switzerland) was dissolved in PBS and injected of 62.5 µg/kg. Spectrofluorometric measurement of the Visudyne distribution was performed at different time points using an optical fiberbased spectrofluorometer. The detected Visudyne signal was analyzed by an imaging spectrometer and analyzed by computer. The mean fluorescence signal after removing the underlying autofluorescence was plotted for three areas in each animal.
Lung function
Mice were anesthetized, after which a small incision was made to expose the trachea, and a cannula was inserted to connect to an inline nebulizer and ventilator. Mice were then challenged with aerosolized PBS followed by increasing doses of methacholine (Sigma-Aldrich). Airway resistance was determined by analysis of pressure and flow waveforms.
Western blot
The protein from the mouse lung or cultured cells was extracted using RIPA lysis buffer (1% NP40, 0.1% Sodium dodecyl sulfate (SDS), 100 μg/ml phenylmethylsulfonyl fluoride, 0.5% sodium deoxycholate, in PBS) on ice. The supernatants were collected after centrifugation at 12,000×g at 4°C for 20 min. Protein concentration was determined using a BCA protein assay kit (Bio-rad, China), and whole lysates were mixed with 4× SDS loading buffer (125 mmol/l Tris-HCl, 4% SDS, 20% glycerol, 100 mmol/l Dithiothreitol (DTT), and 0.2% bromophenol blue) at a ratio of 1:3. Samples were heated at 100°C for 5 min and were separated on SDS-polyacrylamide gels. The separated proteins were then transferred to a PVDF membrane. The membrane blots were first probed with a primary antibody. After incubation with horseradish peroxidase-conjugated second antibody, autoradiograms were prepared using the enhanced chemiluminescent system to visualize the protein antigen. The signals were recorded using X-ray film. Primary antibodies were anti-tumor necrosis factor α (TNFα), anti-IL6, anti-CXCL1, anti-VEGFa, anti-VEGFR2 and α-tubulin (all purchased from Cell Signaling, St Louis, MO, USA). α-tubulin was used as a protein loading control. Secondary antibody is HRP-conjugated anti-rabbit (Jackson ImmunoResearch Labs, West Grove, PA, USA). Images shown in the figure were representative from 5 repeats. Densitometry of Western blots was quantified with NIH ImageJ software (Bethesda, MA, USA). The protein levels were first normalized to loading controls, and then normalized to experimental controls.
Statistical analysis
All statistical analyses were carried out using the SPSS 18.0 statistical software package. All values are depicted as mean ± standard deviation from 5 individuals and are considered significant if p < 0.05. All data were statistically analyzed using one-way ANOVA with a Bonferroni correction, followed by a Fisher's exact test, as necessary.
Results
ACE2 does not alter VEGFa levels in ALI mouse lung
The mouse bleomycin-ALI model has been performed as has been described before. As a routine quality control, we isolated protein from the lung of the mice treated with bleomycin only (ALI) and ACE2 after bleomycin (ALI + ACE2). We then analyzed the inflammatory cytokine levels by Western blot, compared to the mice without treatment (UT). We found that ALI induced significantly increases in TNFα, IL6 and CXCL1 levels, by quantification (Fig. 1A), and by representative images (Fig. 1B). These data confirmed the successful establishment of mouse ALI model by bleomycin and the therapeutic effects of ACE2 in bleomycin-induced ALI. Then, we examined the effects of VEGFa and VEGFR2, and we found that the levels of VEGFa and VEGFR2 in mouse lung were significantly increased by bleomycin treatment, but ACE2 did not altered VEGFa and VEGFR2 levels.

ACE2 does not alter VEGFa levels in ALI mouse lung. We isolated protein from the lung of the mice treated with bleomycin only (ALI) and ACE2 after bleomycin (ALI + ACE2). We then analyzed the inflammatory cytokine levels by Western blot, compared to the mice without treatment (UT). (A-B) The levels of TNFα, IL6, CXCL1, VEGFa and VEGFR2 were analyzed, shown by quantification (A), and by representative images (B). *p < 0.05. NS: non-significant. N = 10.
ACE2 does not alter VEGFa levels in ALI mouse lung. We isolated protein from the lung of the mice treated with bleomycin only (ALI) and ACE2 after bleomycin (ALI + ACE2). We then analyzed the inflammatory cytokine levels by Western blot, compared to the mice without treatment (UT). (A-B) The levels of TNFα, IL6, CXCL1, VEGFa and VEGFR2 were analyzed, shown by quantification (A), and by representative images (B). *p < 0.05. NS: non-significant. N = 10.
ACE2 treatment attenuates ALI-induced increases in lung vessel permeability
Next, we examined the effects of ACE2 on lung vessel permeability at 0 day, 3 days and 7 days after ACE2 treatment. The fluorescence pharmacokinetics profile of the photosensitizer was measured 15 minutes after i.v. injection of 62.5 µg/kg Visudyne. We found that ALI significantly increased lung vessel permeability, while ACE2 alleviated the ALI-induced increases in lung vessel permeability (Fig. 2). Since VEGFa is a major trigger for increasing vessel permeability and ACE2 did not alter VEGFa levels, these data suggest that ACE2 may antagonize the effects of VEGFa-mediated increases in lung vessel permeability.
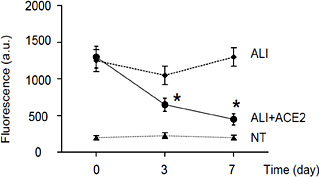
ACE2 treatment attenuates ALI-induced increases in lung vessel permeability. We examined the effects of ACE2 on lung vessel permeability at 0 day, 3 days and 7 days after ACE2 treatment. The fluorescence pharmacokinetics profile of the photosensitizer was measured 15 minutes after i.v. injection of 62.5 µg/kg Visudyne. We found that ALI significantly increased lung vessel permeability, while ACE2 alleviated the ALI-induced increases in lung vessel permeability. *p < 0.05. N = 10.
ACE2 treatment attenuates ALI-induced increases in lung vessel permeability. We examined the effects of ACE2 on lung vessel permeability at 0 day, 3 days and 7 days after ACE2 treatment. The fluorescence pharmacokinetics profile of the photosensitizer was measured 15 minutes after i.v. injection of 62.5 µg/kg Visudyne. We found that ALI significantly increased lung vessel permeability, while ACE2 alleviated the ALI-induced increases in lung vessel permeability. *p < 0.05. N = 10.
Ectogenic VEGFa abolishes the antagonizing effects of ACE2 on the vessel permeability against VEGFa
In order to confirm that ACE2 may antagonize the effects of VEGFa-mediated increases in lung vessel permeability, we implanted a releasing pump of VEGFa at the time of ACE2 treatment (ALI + ACE2 + VEGFa pump). Mini-pumps containing PBS only were implanted in control mice (ALI + ACE2 + CTL pump). We found that provision of ectogenic VEGFa abolished the antagonizing effects of ACE2 on the vessel permeability against VEGFa (Fig. 3A), and then the effects of ACE2 on lung function protection in a methacholine response test for lung resistance (Rl) (Fig. 3B). Hence, this loss-of-function experiment suggests that ectogenic VEGFa may abolish the antagonizing effects of ACE2 on the vessel permeability against VEGFa, resulting in aggravated lung function.

Ectogenic VEGFa abolishes the antagonizing effects of ACE2 on the vessel permeability against VEGFa. We implanted a releasing pump of VEGFa at the time of ACE2 treatment (ALI + ACE2 + VEGFa pump). Mini-pumps containing PBS only were implanted in control mice (ALI + ACE2 + CTL pump). (A) Provision of ectogenic VEGFa abolished the antagonizing effects of ACE2 on the vessel permeability against VEGFa. (B) The effects of ACE2 on lung function protection in a methacholine response test for lung resistance (Rl). *p < 0.05. N = 10.
Ectogenic VEGFa abolishes the antagonizing effects of ACE2 on the vessel permeability against VEGFa. We implanted a releasing pump of VEGFa at the time of ACE2 treatment (ALI + ACE2 + VEGFa pump). Mini-pumps containing PBS only were implanted in control mice (ALI + ACE2 + CTL pump). (A) Provision of ectogenic VEGFa abolished the antagonizing effects of ACE2 on the vessel permeability against VEGFa. (B) The effects of ACE2 on lung function protection in a methacholine response test for lung resistance (Rl). *p < 0.05. N = 10.
Suppression of VEGF signaling mimics the effects of ACE2 on the vessel permeability against VEGFa
In a gain-of-function experiment, we implanted a releasing pump of a VEGF signaling inhibitor, SU5416, after induction of ALI (ALI + SU5416 pump). Mini-pumps containing PBS only were implanted in control mice (ALI + CTL pump). We found that provision of VEGF inhibitor SU5416 mimicked the effects of ACE2 on the vessel permeability against VEGFa (Fig. 4A), and then the effects of ACE2 on lung function protection in a methacholine response test for RI (Fig. 4B). Hence, these data suggest that suppression of VEGF signaling may mimic the effects of ACE2 on the vessel permeability against VEGFa, resulting in improved lung function. Together, our data suggest that ACE2 may antagonize the VEGFa-mediated increases in lung vessel permeability during ALI, resulting in improvement of lung function after ALI.
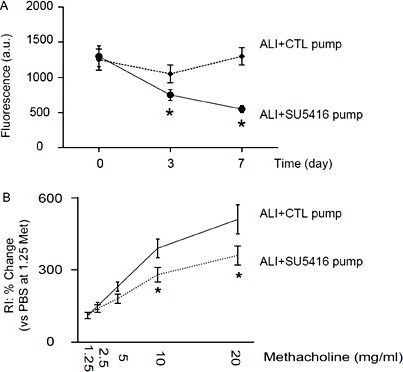
Suppression of VEGF signaling mimics the effects of ACE2 on the vessel permeability against VEGFa. We implanted a releasing pump of a VEGF signaling inhibitor, SU5416, after induction of ALI (ALI + SU5416 pump). Mini-pumps containing PBS only were implanted in control mice (ALI + CTL pump). (A) Provision of VEGF inhibitor SU5416 mimicked the effects of ACE2 on the vessel permeability against VEGFa. (B) The effects of ACE2 on lung function protection in a methacholine response test for RI. *p < 0.05. N = 10.
Suppression of VEGF signaling mimics the effects of ACE2 on the vessel permeability against VEGFa. We implanted a releasing pump of a VEGF signaling inhibitor, SU5416, after induction of ALI (ALI + SU5416 pump). Mini-pumps containing PBS only were implanted in control mice (ALI + CTL pump). (A) Provision of VEGF inhibitor SU5416 mimicked the effects of ACE2 on the vessel permeability against VEGFa. (B) The effects of ACE2 on lung function protection in a methacholine response test for RI. *p < 0.05. N = 10.
Discussion
ALI and ARDS are severe forms of diffuse lung disease that impose a substantial health burden worldwide each year. Data from several population-based studies reveals a fairly consistent picture for the age, mortality, severity of illness, ratio of ARDS to ALI, and ratio of ALI to acute respiratory failure, yet there is almost a four-fold difference in the reported incidence of ALI/ARDS between studies [1,2,3,4]. The resolution from lung injury is not simply relief from injurious agents or factors, but rather reflects an actively regulated program involving removal of apoptotic neutrophils, remodeling of matrix, resolution of protein rich alveolar fluid, and engagement of numerous signaling pathways distinct from those involved in acute injury. Type II alveolar epithelial cells proliferate to cover the injured basement membrane and differentiate into the type I alveolar epithelial cells. Apoptosis is thought to be a major mechanism for the clearance of neutrophils from the injured lung. Among all these processes that may alleviate the severity of ALI, decreasing the lung vessel permeability appears to be a critical ones, since all inflammatory cells, pro-inflammatory cytokines and other factors that trigger the pulmonary pathological changes stem from the increases in lung vessel permeability.
The protective roles of ACE2 in acute and chronic lung diseases have been demonstrated, including anti-inflammation and anti-oxidation [20,21,22,23,25]. In addition, we recently reported a role of ACE2 in inhibiting cell apoptosis after ALI [24,26]. However, the effects of ACE2 on vessel permeability and its relationship with VEGFa remain ill-defined.
Here, we used a previously published bleomycin method to induce ALI in mice, and treated the mice with ACE2. We then analyzed the levels of VEGFa in these mice. The mouse lung vessel permeability was determined by a fluorescence pharmacokinetic assay following i.v. injection of Visudyne. We found that ACE2 treatment did not alter VEGFa levels in lung, but antagonized the effects of VEGFa on increases of lung vessel permeability. These effects may be conducted through competition on the modulation of intracellular downstream factors that regulate the changes in the endothelial-endothelial cell junctions, but not at the levels of receptor binding, since the VEGFa levels were not altered during ACE2 treatment. Future studies may address the intracellular functional factor for this regulation.
In our previous study, we found that ALI induced phosphorylation of a key factor SMAD2 of TGFβ receptor signaling [29,30,31], which could be dose-dependently inhibited by ACE2. Moreover, ACE2-induced suppression of miR-4262 induced the inhibition of the PEC apoptosis after ALI through Bcl-2. Together with the data shown here, we have now demonstrated that ACE2 may have its therapeutic effects on ALI through different molecular signaling pathways. These studies may contribute to our understanding of using ACE2 as a potent treatment for curing ALI and for preventing ARDS.
Disclosure Statement
The authors have declared that no conflict of interest exists.

