

Abstract
Congenital hyperinsulinism (CHI), characterized by profound hypoglycaemia related to inappropriate insulin secretion, may be associated histologically with either diffuse insulin hypersecretion or focal adenomatous hyperplasia, which share a similar clinical presentation, but result from different molecular mechanisms. Whereas diffuse CHI is of autosomal recessive, or less frequently of autosomal dominant, inheritance, focal CHI is sporadic. The most common mechanism underlying CHI is dysfunction of the pancreatic ATP-sensitive potassium channel (K+ATP). The two subunits of the K+ATP channel are encoded by the sulfonylurea receptor gene (SUR1 or ABCC8) and the inward-rectifying potassium channel gene (KIR6.2 or KCNJ11), both located in the 11p15.1 region. Germ-line, paternally inherited, mutations of the SUR1 or KIR6.2 genes, together with somatic maternal haplo-insufficiency for 11p15.5, were shown to result in focal CHI. Diffuse CHI results from germ-line mutations in the SUR1 or KIR6.2 genes, but also from mutations in several other genes, namely glutamate dehydrogenase (with associated hyperammonaemia), glucokinase, short-chain L-3-hydroxyacyl-CoA dehydrogenase, and insulin receptor gene. Hyperinsulinaemic hypoglycaemia may be observed in several overlapping syndromes, such as Beckwith-Wiedemann syndrome (BWS), Perlman syndrome, and, more rarely, Sotos syndrome. Mosaic genome-wide paternal isodisomy has recently been reported in patients with clinical signs of BWS and CHI. The primary causes of CHI are genetically heterogeneous and have not yet been completely unveiled. However, secondary causes of hyperinsulinism have to be considered such as fatty acid oxidation deficiency, congenital disorders of glycosylation and factitious hypoglycaemia secondary to Munchausen by proxy syndrome.
Introduction
Congenital hyperinsulinism (CHI, MIM 256450) related to inappropriate insulin secretion is the most common cause of hypoglycaemia in newborns and infants. Hypoglycaemia revealed by seizures in about half of the cases is often severe with secondary brain damage [1,2,3,4]. Although the clinical presentation of hyperinsulinism is similar, it may result from different molecular causes [5,6,7,8,9,10].
The diagnostic criteria for CHI include recurrent fasting and fed hypoglycaemia (<3 mmol/l) with inadequate insulin plasma levels requiring high rates of intravenous (IV) glucose (>10 mg/kg/min) and increased plasma glucose after IV glucagon injection. In the absence of clearly abnormal insulin levels during the hypoglycaemic episode, a 4- to 6-hour fasting test in search of inappropriately low plasma levels of ketone bodies, free fatty acids, and branched chain amino acids, may be helpful. Mild hepatomegaly is common and does not prevent the diagnosis of hyperinsulinism. Facial dysmorphism with high forehead, large and bulbous nose with short columella, smooth philtrum and thin upper lip is frequently observed in all types of hyperinsulinism [11]. Epilepsy seems to be frequent in patients with hyperinsulinism associated to hyperammonaemia and is not explained by hypoglycaemia only [12]. However, mostly, hypoglycaemia is the only symptom.
The onset of hypoglycaemia is reported to occur in the neonate or after the first month of life [13,14,15]. In the neonatal period, hypoglycaemia is severe (often <1 mmol/l) and occurs within the first 72 h of life. The majority of affected newborns with severe CHI are macrosomic at birth. Other symptoms are: abnormal movements, tremulousness, hypotonia, cyanosis, or hypothermia. In some cases, hypoglycaemia is discovered by routine measurement of blood glucose. CHI patients presenting with hypoglycaemia later in infancy (1–12 months of age) have a similar clinical presentation, but usually require lower rates of IV glucose to maintain glycaemia within normal ranges.
CHI may be associated histologically with two major forms: diffuse insulin hypersecretion or focal adenomatous hyperplasia. Both forms share a similar clinical presentation. Histologically, focal adenomatous hyperplasia is a small poorly delineated lesion composed of normally structured hyperplastic islets (β cells surrounded by non-β cells), separated by few exocrine acini, thus maintaining a normal lobular pancreatic architecture [16]. A high proliferation rate of β cells was shown inside the lesion, whereas in the normal adjacent pancreas, small resting islets, made of packed endocrine cells with scanty cytoplasm, exhibit no sign of proliferation [2]. Furthermore, loss of the maternally expressed CDKN1C gene within the lesion is evidenced by the absence of immunohistochemical staining of the corresponding protein, in contrast to normal surrounding islets [16, 17]. These features differ from true adult-type pancreatic adenoma or insulinoma which are microscopically less well distinguished from adjacent pancreatic tissue, sometimes extending between exocrine acini or including normal islets. Outside insulinomas, islets show regular nuclei and normally abundant cytoplasm and are thus very different from those located in non-lesional pancreas of focal forms [16, 17]. Diffuse hyperinsulinism is characterized histologically by the presence of β cells with a particularly abundant cytoplasm and a large nucleus in the islets throughout the whole pancreas [18,19,20,21]. These features are interpreted as a morphological evidence for the continuous hyperfunction of β cells [18, 19, 22].
Clinical features and preoperative classical radiology of the pancreas, including sonography, CT scan and MRI, cannot discriminate between focal and diffuse disease. Therefore, pancreatic venous sampling and pancreatic arterial calcium stimulation were, until recently, the only preoperative procedures available for localizing the site of insulin secretion [23, 24]. Pancreatic venous sampling allows to collect venous blood samples from the entire pancreas (head, isthmus, body and tail) for measurements of plasma glucose, insulin and C-peptide levels [23]. Patients with a focal lesion have high plasma insulin and C-peptide levels in one or more consequent samples, and low plasma insulin and C-peptide levels in the remaining pancreatic samples. By contrast, patients with diffuse hyperinsulinism have high plasma insulin and C-peptide levels in all pancreatic samples [23, 24]. A new accurate non-invasive technique, [18F]-fluoro-L-dopa whole-body PET, has only recently become available to detect hyperfunctional pancreatic islets. Abnormal focal uptake of [18F]-fluoro-L-dopa is observed in the pancreas of patients with a focal lesion, while a diffuse uptake of the radiotracer is observed over the whole pancreas for patients with diffuse insulin secretion [25, 26]. PET scanning with 18F–dopa is to date the most employed method to distinguish between focal and diffuse CHI.
The treatment of hyperinsulinemic hypoglycaemia must be rapid and aggressive in order to prevent irreversible brain damage. In neonates, this often necessitates central venous access and continuous oral feeding using a nasogastric tube. Glucagon (1–2 mg/day given as a continuous subcutaneous infusion) can be added if blood glucose levels remain unstable despite a high glucose infusion rate.
Specific treatments must also be started concomitantly: diazoxide at 15 mg/kg/day in neonates and 10 mg/kg/day in infants, given orally 3 times a day. Diazoxide efficacy is defined as a normalization of blood glucose levels (>3 mmol/l) measured before and after each meal in patients with physiological feeding and after stopping IV glucose and any other medications for at least 5 consecutive days. Diazoxide is usually effective in the infantile form, but most patients with the neonatal form are resistant to this treatment [27]. In case of non-responsiveness to diazoxide, octreotide can be tried at 10–50 µg/d, given either in 3–4 subcutaneous (SC) injections or by SC pump. Other drugs such as calcium-channel blockers (e.g. nifedipine) have been proposed, but are rarely efficient.
Hyperinsulinism associated with hyperammonaemia (HA/HI syndrome) is usually amenable to diazoxide or a restricted protein diet (limiting the leucine intake to 200 mg per meal).
Secondary causes of hyperinsulinism should also be excluded, namely fatty acid oxidation defects (acylcarnitine profile), congenital disorders of glycosylation (transferrin glycosylation), and Munchausen syndrome by proxy [28], as the treatment will be different. Figure 1 contains a decision tree that may be useful for the clinical management of patients with CHI.

Decision tree for clinical management of patients with CHI. Hyperammonaemia is found in HA/HI syndrome. Methylation abnormalities at 11p15 region can be found in syndromic forms of hyperinsulinism, namely in BWS. Secondary causes of hyperinsulinism such as fatty acid oxidation defects detected by acylcarnitine profile, and congenital disorders of glycosylation detected by transferrin glycosylation studies have to been excluded. NH3 = Ammonaemia; acylcarn. = acylcarnitine profile; transferrin = transferrin glycosylation; meth. 11p15 = detection of methylation abnormalities at 11p15 region; dom. = autosomal dominant inheritance.
Decision tree for clinical management of patients with CHI. Hyperammonaemia is found in HA/HI syndrome. Methylation abnormalities at 11p15 region can be found in syndromic forms of hyperinsulinism, namely in BWS. Secondary causes of hyperinsulinism such as fatty acid oxidation defects detected by acylcarnitine profile, and congenital disorders of glycosylation detected by transferrin glycosylation studies have to been excluded. NH3 = Ammonaemia; acylcarn. = acylcarnitine profile; transferrin = transferrin glycosylation; meth. 11p15 = detection of methylation abnormalities at 11p15 region; dom. = autosomal dominant inheritance.
Patients resistant to medical treatment require pancreatectomy: focal CHI can be definitively cured by a limited pancreatectomy, while diffuse CHI requires a subtotal pancreatectomy, with a high risk of secondary diabetes mellitus. Intraoperative histological analysis is performed to provide confirmation of the findings of pancreatic catheterization or PET scanning and to guide the limits of pancreatic resection, in patients suspected of focal CHI.
Molecular Basis
The most common mechanism underlying CHI is dysfunction of the pancreatic ATP-sensitive potassium channel (K+ATP). The two subunits of theK+ATP channel are encoded by the sulfonylurea receptor gene (SUR1 or ABCC8) and the inward-rectifying potassium channel gene (KIR6.2. or KCNJ11), both located in the 11p15.1 region. K+ATP channels are open in unstimulated β cells, establishing a resting membrane potential of –65 mV. At this state, the intracellular ATP/ADP ratio is low (ATP 1 mmol/l, ADP 40 mmol/l), keeping the channels, which have a high sensitivity to ADP, open. Following glucose uptake and metabolism, ATP/ADP increases, ADP concentration decreases, and leads to closing of K+ATP channels and depolarization of the cell membrane. This opens the voltage-gated Ca2+ channels, allows the influx of extracellular calcium, and the exocytosis of insulin. CHI ‘channelopathies’ are due to inhibiting SUR1 or KIR6.2 mutations. These mutations can lead to type 1 channelopathy without channel activity, or to type 2 channelopathy with a decreased channel activity due either to defective function, or to decreased number of channels. Heterogeneous outcome is observed for the same mutation as some cells manifest a type 1 channelopathy, others a type 2 channelopathy, and other mutated cells have a normal activity of the potassium channel [29, 30]. This observation could be explained by interactions with modulator genes, exogenous factors, or variable degree of penetrance of the mutation.
Mutations of SUR1 gene are responsible for 50–60% of CHI, whatever focal or diffuse CHI, especially in neonates. More than 100 distinct mutations, distributed throughout the SUR1 gene, have already been described [31,32,33,34]. Mutations of KIR6.2 gene are less frequent and are responsible for 10–15% of CHI.
Less frequent mechanisms of CHI, observed especially in infants, and responsible for diffuse CHI, the ‘metabolopathies’, are due to enzyme deficiencies of glutamate dehydrogenase (GDH), glucokinase (GK), short-chain L-3-hydroxyacyl-CoA dehydrogenase (SCHAD) or to insulin receptor dysfunctions (fig. 2). In contrast to patients with ‘channelopathies’, those with ‘metabolic’ CHI are sensitive to diazoxide (which acts on the potassium channel), as K+ATP channels are functional in these patients.
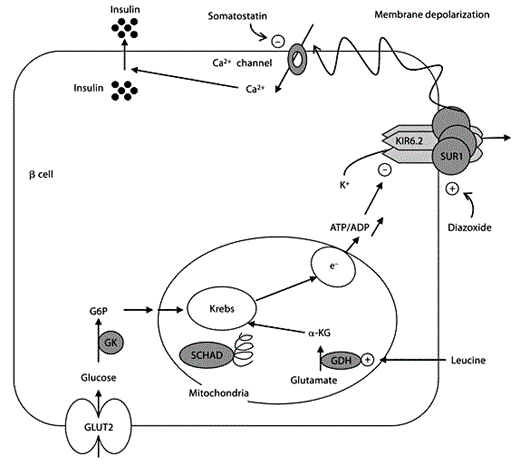
Schematic representation of the regulation of insulin secretion by glucose in pancreatic βcells. GLUT2 catalyses glucose uptake by β cells. The phosphorylation of glucose to glucose-6-phosphate by glucokinase initiates β-cell glucose metabolism. Leucine, one of the most potent amino acids in stimulating insulin secretion, acts indirectly as a positive allosteric effector of glutamate dehydrogenase increasing the rate of oxidation of glutamate to α-ketoglutarate. Both glucose and leucine interact with the Krebs cycle activity resulting in ATP synthesis. This increase of the ATP/ADP ratio triggers the closure of the potassium channel, leading to depolarization of the cell membrane, influx of extracellular calcium, and release of insulin from storage granules. The several pathways involved in insulin secretion explain the modality of effectiveness of medical cures such as diazoxide, somatostatin, and protein-restricted diet. GLUT2 = Glucose transporter 2; G6P = glucose-6-phosphate; GK = glucokinase; GDH = glutamate dehydrogenase; α-KG = α-ketoglutarate; Krebs = Krebs cycle; e– = mitochondrial respiratory chain; SUR1 = sulfonylurea receptor; KIR6.2 = inward-rectifying potassium channel are the subunits of potassium channel.
Schematic representation of the regulation of insulin secretion by glucose in pancreatic βcells. GLUT2 catalyses glucose uptake by β cells. The phosphorylation of glucose to glucose-6-phosphate by glucokinase initiates β-cell glucose metabolism. Leucine, one of the most potent amino acids in stimulating insulin secretion, acts indirectly as a positive allosteric effector of glutamate dehydrogenase increasing the rate of oxidation of glutamate to α-ketoglutarate. Both glucose and leucine interact with the Krebs cycle activity resulting in ATP synthesis. This increase of the ATP/ADP ratio triggers the closure of the potassium channel, leading to depolarization of the cell membrane, influx of extracellular calcium, and release of insulin from storage granules. The several pathways involved in insulin secretion explain the modality of effectiveness of medical cures such as diazoxide, somatostatin, and protein-restricted diet. GLUT2 = Glucose transporter 2; G6P = glucose-6-phosphate; GK = glucokinase; GDH = glutamate dehydrogenase; α-KG = α-ketoglutarate; Krebs = Krebs cycle; e– = mitochondrial respiratory chain; SUR1 = sulfonylurea receptor; KIR6.2 = inward-rectifying potassium channel are the subunits of potassium channel.
Modes of Inheritance
Sporadic Forms
Recent estimates from France, Japan and the United States suggest that 40–65% of all CHI patients have a focal form [31, 35, 36].
Focal CHI has been shown to result from a paternally inherited mutation in the SUR1 or KIR6.2 genes and loss of the maternal 11p15 allele (loss of heterozygosity, LOH). LOH is a somatic event restricted to the pancreatic lesion which leads to tumour inception through disruption of the balance of expression of several imprinted genes located in the 11p15.5 region and controlling cell growth [37]. Because of the somatic character of LOH, focal CHI is a sporadic event. In patients with focal CHI, the pancreas lesion is generally unique with a size of <10 mm in the largest aspect [16,18,19,20,38,39,40]. Rarely, patients with multiple-focal lesions or giant lesions have been reported [38]. The underling molecular mechanism of these forms is similar to the one described in the small solitary forms but the onset during the pancreas embryogenesis may be different: earlier for the giant forms and multiple-hit for the multiple-focal forms [41].
Patients with de novo germ-line mutations in genes responsible for diffuse forms of CHI may be considered initially as sporadic cases.
Autosomal Recessive Inheritance
Autosomal Dominant Inheritance
The second most common form of CHI involving the GLUD1 gene, coding for GDH, is often associated with hyperammonaemia (HI/HA syndrome) [43]. Dominantly expressed missense mutations of GLUD1 gene result in a gain of function of GDH, a mitochondrial matrix enzyme. This induces an increase of the oxidative deamination of glutamate in α-ketoglutarate and ammonium, responsible for an increased Krebs cycle activity, which results in an increased ATP/ADP ratio, and consequently activation of K+ATP channel with subsequent cell depolarization and insulin release [9].
Dominantly expressed GK mutations are a rare cause of CHI [8]. They result in a gain of function by increased affinity of GK for glucose leading to inappropriate insulin secretion. These mutations are remote from the glucose-binding site and suggest an allosteric regulation defect.
Less frequently than in the recessive (diffuse CHI) or sporadic (focal CHI) form, SUR1 gene can also be involved in dominant CHI. In this case, the histological lesion is diffuse.
The insulin receptor (INSR) gene was recently implicated in a dominant form of hyperinsulinemic hypoglycaemia [44]. Patients presented with postprandial as well as fasting hyperinsulinemic hypoglycaemia associated with resistance to insulin [44].
Exercise-induced hyperinsulinism is a novel, autosomal dominant form of CHI, which has been identified in two families. The patients suffer from hypoglycaemic symptoms only when performing strenuous physical exercise [45]. The underlying mechanism of hypoglycaemia is unknown, so far.
Syndromic Forms
Finally, hyperinsulinemic hypoglycaemia can be ‘syndromic’ as observed in several overlapping syndromes, such as Beckwith-Wiedemann syndrome (BWS) [46], Perlman syndrome [47] and, more rarely, in Sotos syndrome [48]. BWS results from several identified genetic and epigenetic molecular events including paternal isodisomy [49], abnormal methylation of IGF2/H19 [50], chromosomal aberrations involving the 11p15 region [51], and CDKN1C mutation [52]. Hypoglycaemia in BWS patients has been associated with paternal uniparental disomy of 11p15 rather than other genetic abnormalities [46], but the pathophysiological mechanism leading to hyperinsulinic hypoglycaemia is still unclear as no evidence for duplication of INS, HRAS1 and IGF2 [53] or overexpression of the INS and IGF2 genes [54] was found. A case of BWS with CHI and mosaic genome-wide paternal isodisomy has been reported [55], and we may suggest that mosaic and genome-wide paternal isodisomy are likely to be underdiagnosed in patients with clinical signs of BWS or CHI.
The genetic mechanism of CHI and the most frequent modes of inheritance are summarized in table 1.

Conclusion
CHI involves widely heterogeneous genes and mechanisms. Other genes encoding transcription factors as well as genes implicated in β-cell metabolism are probably involved. Furthermore, CHI association with disorders known to be related to imprinted regions of the human genome should be the focus of further in-depth diagnostic efforts.
