

Abstract
Whole-genome sequencing was performed on 3 bipolar I disorder (BPI) cases from a multiplex pedigree of European ancestry with 7 BPI cases. Within CACNA1D, a gene implicated by genome-wide association studies, a G to C nucleotide transversion at 53,835,340 base pairs (bps) was found predicting the substitution of proline for alanine at amino acid position 1751 (A1751P). Using Sanger sequencing, the DNA variant was shown to co-segregate with the remaining 4 BPI cases within the pedigree. A high-resolution DNA denaturing curve method was then used to screen for the presence of the A1751P change in 4,150 BPI cases from the NIMH Genetics Initiative. The A1751P variant was found in 4 BPI cases. A second variant within exon 43, a C to T nucleotide transition, was found in 1 case at 53,835,355 bps, predicting the substitution of tryptophan for arginine at amino acid position 1771 (R1771W). In the NHLBI Exome Sequencing Project database, the heterozygous A1751P variant was present in 3 of 4,300 subjects of European ancestry, and the R1771W change was not present in any subject. Given the rarity of these variants, large-scale case/control rare variant sequencing studies will be required for definitive conclusions.
Introduction
Bipolar I disorder (BPI), also termed manic depressive illness, is a genetically based complex neuropsychiatric disorder that afflicts almost 1% of the world's population [1]. Genome-wide linkage analyses of multiplex pedigrees have not produced definitive findings, but results are consistent with a wide range of locus heterogeneity for disease etiology [2]. A few studies have reported evidence of linkage between BPI and the 3p region that contains the calcium channel, voltage-dependent, L type, alpha 1D subunit gene (CACNA1D) [3,4,5]. Genome-wide association studies (GWAS) of BPI have yielded several statistically significant findings, although the implicated loci explain only a fraction of the genetic variation [6,7]. Of interest, a number of calcium channel genes have been implicated by GWAS of BPI as well as schizophrenia and autism [8,9]. Association analysis of 11,974 BPI cases and 51,792 controls yielded statistically significant findings with CACNA1C [6]. Evidence of association - though not genome-wide significant - was also found with CACNB3 (p = 2.51 × 10−6) and CACNA1D (p = 3.73 × 10−5).
In addition to common variants, rare variants are believed to also play a significant role in BPI and other major psychiatric disorders. The recent development of relatively low-cost whole-exome and whole-genome sequencing [10,11] has now made it possible to perform rare variant association studies using large case/control samples. In the first relatively large study of schizophrenia, whole-exome sequencing of 2,500 schizophrenia cases and 2,500 controls did not yield statistically significant evidence of association with any one gene [12], suggesting that there may be hundreds if not thousands of disease loci. The genetics of BPI is likely to be no less complicated. Given this heterogeneity, it is not surprising that previous linkage studies were inconclusive - although evidence of major loci was found in a few exceptionally large pedigrees. In order to discover rare loci underlying BPI via whole-exome or whole-genome sequencing, two types of sampling strategies are available: case/control or large multiplex families. Given the large range of heterogeneity, very large case/control samples will be required - perhaps up to 100,000 cases and 100,000 controls [13]. While not currently feasible, with the future reduction of sequencing costs coupled with continued ascertainment of cases, such a study will likely be carried out in the future [14]. In the meantime, family-based sequencing approaches can be carried out at relatively low cost using uniquely large pedigrees that may already be in hand, many having been used in previous linkage studies. In a multiplex family, only one locus (monogenetic transmission) or only a few disease loci (oligogenetic inheritance) may be operating. While the contribution of family-based sequencing studies to rare variant identification will be modest - given the limited number of multiplex families in the population - they complement case/control studies. For instance, many, if not most, of the loci found in families will also be implicated in case/control studies. Some of the loci discovered in case/control sequencing should also be found to segregate in families. Showing co-segregation in a family provides additional evidence that a susceptibility gene has been discovered.
In this study, a multiplex pedigree of European ancestry, K1561, containing 7 BPI cases was ascertained. Of the 7 cases, 6 are sibs and 1 case is the offspring of an affected sib (fig. 1). The average age of illness onset is 27.6 years (range 16-48). Six of the 7 BPI cases were treated with lithium and all were responsive. The other BPI case was successfully treated with antipsychotics. None of the manic episodes appeared to have been induced by treatment with antidepressants. The family includes 2 cases with BPII and 2 individuals with recurrent major depression, one with DNA available. The sister of the father of the BPI sibs has recurrent major depression. There is no family history of mood disorders on the maternal side. Whole-genome sequencing of 3 BPI sibs was carried out. Of 115 rare variants identified by annotation in 113 genes, one was in CACNA1D, a calcium channel subunit gene located on chromosome 3p14.3 that has been implicated by GWAS.
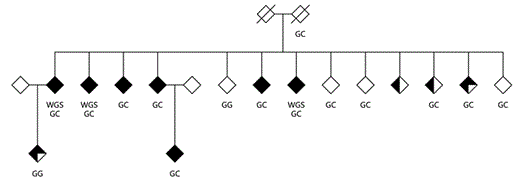
K1561 showing 7 BPI, 2 BPII and 2 recurrent unipolar depression cases. The fully filled in diamonds are BPIs, three-quarter filled BPIIs and half filled recurrent major depression. The subjects' whole genome sequences are denoted by WGS. The pedigree was ascertained via the oldest BPI individual in the second generation. Of the 7 BPI cases, 6 have children: two with four each, three with two each and one with three.
K1561 showing 7 BPI, 2 BPII and 2 recurrent unipolar depression cases. The fully filled in diamonds are BPIs, three-quarter filled BPIIs and half filled recurrent major depression. The subjects' whole genome sequences are denoted by WGS. The pedigree was ascertained via the oldest BPI individual in the second generation. Of the 7 BPI cases, 6 have children: two with four each, three with two each and one with three.
Methods
This study was approved by the UCSF Committee on Human Research, San Francisco, Calif., USA. After informed consent, all family members of K1561 were interviewed using a semi-structured instrument, the SADS-L [15], and diagnoses were derived using RDC criteria [16]. DNA from blood was extracted using phenol-chloroform methods. Whole-genome sequencing was contracted out to Complete Genomics that utilized a short-read propriety method [17]. HG19 was used for genome assembly. For the initial annotation, we used the VAT (Variant Annotation Tool) within the Variant Annotation, Analysis and Search Tool (VAAST) package [18]. Identified variants were further analyzed using SIFT (http://sift.jcvi.org), PolyPhen2 (http://genetics.bwh.harvard.edu/pph2/) and Functional Analysis through Hidden Markov Models (FATHMM) [19] using ANNOVAR [20]. Given only modest correlations among the various prediction programs [21], we chose to use a number of methods. We used the following criteria for filtering variants: (1) heterozygous in all 3 cases; (2) potentially functional (stop gain, stop loss, nonsynonymous, inframe indel, frame shift indel, and splice site); (3) allele frequency less than 1% in the 1,000 Genomes project samples, and less than 20% in the Exome Aggregation Consortium (ExAC, http://exac.broadinstitute.org/) samples, and (4) not in a known segmental duplication region. Sanger sequencing was carried out to validate variants and to check for co-segregation with all available individuals in the pedigree of whom four had BPI. A high-resolution melting curve method was used to screen for the presence of the variant in additional samples [22]. A total of 4,153 unrelated BPI cases of European ancestry ascertained by NIMH Genetics Initiative (https://www.nimhgenetics.org) were used to assess the frequency of identified variants. The NHLBI Exome Sequencing Project (ESP) database was used to determine the frequency of variants in a population not known to have major psychiatric disorders (http://evs.gs.washington.edu/EVS/).
Results
For all 3 individuals subjected to whole-genome sequencing, genome coverage averaged at least 30×. The percent of fully called reference bases was ~98.6%, and the falsely heterozygous error rate was less than 1 in 100,000 positions. In total, 5,498,522 variants were identified in the 3 individuals. Annotation using VAT yielded 1,018,290 genic variants that are shared by all 3 cases. Filtering based on genotype (heterozygous in all individuals), annotation (potentially functional), genomic context (not in segmental duplication regions), and population allele frequency (less than 1% in the 1,000 genomes and less than 20% in ExAC) resulted in 115 heterozygous rare potential functional variants in 113 genes shared by all 3 cases (online suppl. table 1, for all online suppl. material, see www.karger.com/doi/10.1159/000448041). Of the 115 variants, one was found in CACNA1D, a G to C nucleotide transversion at 53,835,340 base pairs (bps) in exon 43, predicting the substitution of alanine to proline at amino acid position 1751 (A1751P). This segment is likely to be part of the cytoplasmic terminus. In the VAAST analysis, this nonsynonymous variant scored 38.49 and CACNA1D ranked 24th genome-wide under the dominant mode of inheritance. Neither SIFT nor Polyphen2 predicted this variant to be damaging, but FATHMM denoted the variant as deleterious. The variant co-segregated with the 4 remaining BPI cases. Linkage was carried out with the 7 BPI cases. The nonparametric analysis, as implemented in Merlin, yielded a LOD score of 0.58 (p = 0.05). However, it is possible that the CACNA1D variant co-segregated by chance. Given 115 variants and four meioses separating the 4 additional cases from the 3 affected sibs, one would predict that approximately seven variants would be shared by chance. To assess the frequency of the variant in a large BPI cohort, high-resolution DNA melting curve analyses were performed on 4,153 unrelated BPI cases of European ancestry ascertained by NIMH Genetics Initiative. Of the 4,150 samples that were successfully assayed (three did not yield PCR products), the heterozygous G to C nucleotide transversion was present in 4 cases (4 of 8,300 chromosomes), yielding an allele frequency of 0.00048. All potential variants found by the denaturing analysis were validated by Sanger sequencing. To determine the frequency of the variant in a population not known to have psychiatric disorders, we examined the ESP database. The heterozygous variant was present in 3 of 4,300 subjects (3/8,600 chromosomes) of European ancestry, giving an allele frequency of 0.00035. A second variant was found in the DNA melting curve analyses. Sanger sequencing revealed a heterozygous C to T nucleotide transition at 53,835,355 bps, predicting the substitution of tryptophan for arginine at amino acid position 1756. This variant was not present in the ESP database, and the variant was not found in any of the 3 cases in K1561 that were whole genome sequenced (online suppl. table 1). SIFT predicted the variant to be tolerated, Polyphen2 probably damaging, and FATHMM deleterious. Thus, all together 5 of 4,150 NIMH cases had a rare nonsynonymous variant SNP within this exon, yielding an allele frequency of 0.0006 in the cases versus an allele frequency of 0.00035 in the control ESP database. A χ2 test of the two allele frequencies showed no significant difference between the two groups (χ2 = 0.57, p = 0.45, one degree of freedom, without Yates correlation).
Discussion
Among the three whole-genome sequences, we identified 115 damaging rare variants in 113 genes that were shared by the 3 BPI cases, including a G to C nucleotide transversion in exon 43 of the voltage-gated calcium channel subunit gene CACNA1D located on 3q14. This sequence variant predicts the substitution of proline for alanine at amino acid position 1751. The variant co-segregated with the remaining 4 BPI cases. In addition, the variant was present in 1 of 2 BPII cases, 1 individual with recurrent major depression and 3 unaffected individuals in the pedigree (fig. 1). The presence of the variant in unaffected subjects is expected given reduced penetrance. Imperfect segregation with cases may occur due to misdiagnosis, the presence of a phenocopy, as well as etiologic or genetic heterogeneity. Although neither SIFT nor Polyphen2 denoted the variant to be damaging, FATHMM predicted the variant to be deleterious. Given that annotation programs are at best 70% robust [21], in vitro studies will be required to determine whether the C/T substitution produces functional changes in the calcium channel. The C/T variation was found in 4 of 4,153 BPI cases derived from the NIMH Genetics Initiative. In the ESP database, the variant is present in 3 of 4,300 subjects of European ancestry. A second variant within exon 43, a C to T nucleotide transition, was found at 53,835,355 bps, predicting the substitution of tryptophan for arginine at amino acid position 1751. While SIFT concluded the variant to be tolerated, Polyphen2 denoted it as probably damaging and FATHMM as deleterious. This variant was not present in the ESP database. Although the combined allele frequency for these two variants is higher in cases versus controls, the difference is not significant (χ2, p = 0.45). However, a limitation of the ESP sample is the lack of psychiatric screening; thus, it is possible that some of the subjects with the CACNA1D variants have BPI.
CACNA1D is part of a multi-gene family that encodes the alpha 1 subunit of high-voltage calcium channels. Based on varying pharmacologic and structural properties, high-voltage calcium channels are divided in L-type (dihydropyridine sensitive), P/Q-type (ω-agatoxin sensitive), and N-type calcium channels (selectively inhibited by ω-conotoxins) [23]. Subunits encoded by CACNA1C and CACNA1D are the only L-type subunits that play a significant role in neuronal functioning which includes neurotransmitter release and changes in gene expression [24]. Alterations in calcium signaling have been indirectly implicated in BPI [25]. Calcium channel blockers - which bind to L-type channels - have been used with mixed success in the treatment of BPI, but the clinical trials have all involved a small number of subjects and thus have been underpowered. The calcium channel blockers that are approved for treatment of cardiovascular disease block the alpha 1C subunit - mainly present in the cardiovascular system - at much lower doses than the alpha 1D that is primarily present in the brain (along with the alpha 1C subunit). Moreover, the available calcium channel blockers are not highly lipid soluble. Thus, it is possible that levels sufficient for treatment of BPI were not achieved in these studies. Alterations in circadian rhythms have been reported in BPI, and it is noteworthy that the calcium channel containing the alpha 1D subunit contributes to the spontaneous firing of neurons in the suprachiasmatic nucleus, a brain region that underlies circadian rhythms [26].
CACNA1D comprises 317,462 bps with an open reading frame of 6,546 bps and has 49 exons. We were able to study only one exon, as costs were prohibitive to screen the entire gene in a large cohort. Interestingly, in a recent study where 200 bipolar disorder cases were whole genome sequenced, most of the risk variants predicted in CACNA1D were rare, noncoding promoter and enhancer variants with predicted regulatory effects [27]. To conclusively determine the role of rare variants in CACNA1D in BPI, the whole gene needs to be interrogated using large samples of cases and controls. A recent simulation study by Zuk et al. [13] suggests that a ‘well-powered' association study should use at least 25,000 cases and 25,000 controls, and it is possible that at least 100,000 cases and 100,000 controls will be required. Given the large number of samples required for rare variant association analyses, screening additional multiplex families may be worthwhile. More highly penetrant and biologically relevant risk alleles may be present in families, and family studies can facilitate the identification of disease mechanisms and novel treatments. Therefore, identifying rare variants that segregate in multiplex pedigrees complements case/control association studies and provides a valuable approach for identifying susceptibility genes. Given previous neurobiological treatment and GWAS data, it is perhaps not surprising that CACNA1D might harbor rare variants predisposing to BPI. Nevertheless, results of this study at best provide suggestive data supporting this. Additional work will be required to conclusively determine whether rare variation in CACNA1D plays a role in BPI.
Acknowledgements
We thank Sam Barondes and the UCSF Center for Neurobiology and Psychiatry for funding the initial whole-genome sequencing. We thank Charles Mellon for help in the ascertainment of the pedigree.
Statement of Ethics
This study was approved by the UCSF Committee on Human Research, San Francisco, Calif., USA.
Disclosure Statement
The authors have no conflicts of interest to disclose.
References
For correspondence please contact: Jessica Ross (jessica.ross@ucsf.edu), Jinchuan Xing (xing@biology.rutgers.edu), or William Byerley (william.byerley@ucsf.edu).
