

Abstract
Thyroid cancer derived from follicular cells (TCDFC) comprises well-differentiated (papillary and follicular) carcinoma, poorly differentiated carcinoma, and anaplastic carcinoma. Papillary thyroid carcinoma is the most common endocrine cancer, and its incidence is steadily increasing. Lethality and aggressiveness of TCDFC is inversely correlated with differentiation degree. In this review, an emphasis has been put on molecular markers involved in tumorigenesis of thyroid carcinoma with a focus on aggressive histotypes and the role of such biomarkers in predicting thyroid cancer outcome. Genetic rearrangements in TCDFC (RET/PTC, PAX8/PPARG) and mutations in RAS, BRAF, TERT promoter (TERT p), TP53, PIK3CA, and AKT1 are discussed. The majority of the studies to date indicate that TERT p mutations can serve as a marker of more aggressive disease in all the subtypes of thyroid carcinoma, being the best current marker of poor prognosis, due to its independent association with distant metastases and increased disease-specific mortality. Some studies suggested that a more accurate prediction of thyroid cancer outcome may be possible through a more extensive genetic analysis. The same is true concerning the identification of other mutations that are only relatively frequent in advanced tumors (e.g., TP53, PIK3CA, or AKT1). A better understanding of the prognostic role of TERT p mutation (together with additional ones like BRAF, RAS, PIK3CA, AKT1, or TP53) and the clarification of their putative role in fine-needle aspiration biopsies are likely to allow, in the future, an early refinement of the stratification risk in patients with well-differentiated carcinomas. It is worth noting that, as with any categorical staging system, the risk evaluation within each category (low, intermediate, and high) varies depending on the specific clinicopathologic features of individual patients and the specific biological behavior of the tumor. Finally, besides the diagnostic and/or prognostic significance of the above-mentioned mutations, it is crucial to understand that the molecular pathways and epigenetic alterations will likely turn into interesting targets for new therapies.
Thyroid cancer derived from follicular cells (TCDFC) comprises well-differentiated thyroid carcinoma (WDTC), poorly differentiated, and anaplastic carcinoma. Lethality and aggressiveness of TCDFC is inversely correlated with differentiation degree [Gandolfi et al., 2015].
In this review, a particular emphasis has been put on papillary thyroid carcinoma (PTC), the most frequent subtype of WDTC, and on aggressive histotypes of thyroid cancer. PTC carries a very good prognosis with up to 95% survival after 10 years; despite that, up to 20% of the patients relapse after initial treatment [Handkiewicz-Junak et al., 2010; Davies and Welch, 2014]. Of the patients who relapse, 5-10% do not respond to conventional therapies and may die from the disease. Since most of the WDTC have an indolent behavior, early identification of high-risk patients who will probably have a more aggressive disease is important in order to find a way to prevent the negative evolution of the disease [Shrestha et al., 2015]. The challenge is how to identify patients requiring more aggressive treatment while, at the same time, preserving the majority of the patients from unnecessary treatments and procedures [Esserman et al., 2013; Rosario et al., 2013].
Poorly differentiated thyroid carcinoma (PDTC) occupies an intermediate position between WDTC (PTC and follicular thyroid carcinoma, FTC) and undifferentiated/anaplastic carcinoma (UTC) carrying a significantly worse outcome as compared to WDTC, with a 10-year survival of approximately 50% of the patients [Volante et al., 2004; Haugen et al., 2016]. Diagnostic criteria for PDTC are based on the consensus proposal from Turin and include (after the diagnosis of malignancy based on unequivocal signs of invasiveness) the following 3 features: (1) solid/trabecular/insular microscopic growth pattern, (2) lack of well-developed nuclear features of papillary carcinoma, and (3) one of the following: convoluted nuclei (evidence for partial loss of differentiation in papillary cancer), tumor necrosis, or 3 or more mitoses per 10 high-power fields [Volante et al., 2004, 2007; Asioli et al., 2010a; Haugen et al., 2016].
UTC, although rare (less than 1% of the approximately 63,000 new cases of thyroid cancer in the USA), is the most lethal of the thyroid cancers. The molecular mechanisms responsible for the clinical aggressiveness are not well known [Shi et al., 2015] with mutations of TP53, BRAF, and TERT promoter (TERT p) being the most frequent genetic alterations [Eloy et al., 2015]. Immunohistochemistry is useful for the differential diagnosis between PDTC and UTC. PDTC exhibit diffuse positivity to thyroid transcription factor 1 (TTF1) and focal positivity for thyroglobulin (Tg) while UTCs are by definition negative for Tg and, usually, also for TTF1. PAX8 may be positive in UTC helping to disclose a follicular cell origin of a very anaplastic tumor in the thyroid.
It is believed that during the progression from localized disease to metastatic disease, tumor cells acquire genetic alterations that make them more aggressive [Gandolfi et al., 2015]. The histological features of thyroid cancer (histological subtype, grade of differentiation, presence and extent of necrosis, mitotic index, vascular invasion, extrathyroidal extension) are important prognostic factors. Molecular alterations associated with increased cell proliferation, expression of numerous oncogenes, and loss of several tumor suppressor genes have been explored as additional diagnostic or prognostic indicators in thyroid cancer [for a review, see Tavares et al., 2016]. Several molecular markers have been extensively studied (BRAF, RET/PTC, RAS, PAX8/PPARG, TP53, PIK3CA, AKT1); still the potential associations of these molecular markers with clinical and pathological features of aggressiveness and/or worse prognosis are not entirely consolidated.
In the present study, we review the main genetic alterations present in thyroid carcinomas and discuss their relevance in the assessment of a more accurate prognosis of thyroid cancer patients.
Genetic Changes Involved in Thyroid Tumorigenesis
Genetic Rearrangements in TCDFC
Rearrangements involving the RET proto-oncogene are commonly found in PTC (3-60%) [Nikiforov, 2002; Soares et al., 2011]. The rearrangement takes the form of a constitutive activation of the RET tyrosine kinase domain in the cytoplasm of PTC cells that is able to trigger PTC changes. RET/PTC1 and RET/PTC3 are the most common forms accounting for over 90% of all rearrangements. Both, RET/PTC1 and RET/PTC3, derive from chromosome 10 inversions [Elisei et al., 2012; Tavares et al., 2016].
RET/PTC1 is more frequent in sporadic PTC displaying the classic histotype that occurs in young patients [Soares et al., 2003], whereas RET/PTC3 is more prevalent in the solid variant of PTC, which is prone to display a more aggressive behavior at presentation. The RET/PTC3 rearrangement was frequently found in PTC from children in the setting of the Chernobyl accident [Sugg et al., 1999; Thomas et al., 1999; Nikiforov, 2002; Tavares et al., 2016]. Curiously, RET/PTC rearrangements were also found in the follicular variant of PTC (FVPTC) and in FTC; in the latter, RET/PTC rearrangement is associated with oncocytic tumors with a solid pattern of growth [de Vries et al., 2012]. The prognostic significance of RET/PTC is not yet fully established, but tumors harboring these alterations rarely progress to PDTC and UTC [Tavares et al., 2016].
The PAX8/PPARG rearrangement is most often seen in lesions with follicular growth pattern (FTC and FVPTC) [Marques et al., 2002; Castro et al., 2006; Tavares et al., 2016], detected also in benign lesions (14% of follicular thyroid adenomas, FTA). In some studies, the rearrangement was associated with multifocality and vascular invasion, but there is not sufficient evidence to allow its definition as a prognostic indicator in WDTC [Armstrong et al., 2014; Tavares et al., 2016].
RAS Mutations in TCDFC
The RAS oncogene family regulates 2 important signaling pathways in thyroid cancer, the MAP kinase cascade (Ras-Raf-MEK-ERK) and PI3K/Akt pathway. All 3 RAS genes have been found mutated in thyroid cancers, but mutation of NRAS codon 61 is by far the most frequent, followed by HRAS mutations.
RAS mutations occur both in benign and malignant thyroid tumors: FTA, FTC, PTC, and with variable frequency in PDTC and UTC. They are more frequent in FTC and, when present in PTC, are more prevalent in the FVPTC [Kimura et al., 2003; Nikiforova et al., 2003; Castro et al., 2006; Sobrinho-Simoes et al., 2008]. Higher frequencies of RAS mutations can be found in PDTC and UTC when compared to WDTC [Zhu et al., 2003; Tavares et al., 2016] with prevalences ranging from 18 to 55% and from 4 to 60%, respectively [Basolo et al., 2000; Garcia-Rostan et al., 2003; Quiros et al., 2005; Ricarte-Filho et al., 2009]. Due to the small size and the short follow-up of most series, it is not possible to securely state that RAS mutations per se have prognostic value, although some authors showed increased frequency of distant metastases in patients harboring tumors with RAS mutation [Jang et al., 2014].
BRAF Mutations in TCDFC
A point mutation at codon 600 of the BRAF gene results in the amino acid substitution of a valine by a glutamate (V600E) and was first described in melanoma and then in several types of cancer including thyroid cancer [Davies et al., 2002; Soares et al., 2003; Doherty and Sharma, 2015; Eloy et al., 2015; Gandolfi et al., 2015; Garcia-Rostan et al., 2015; George et al., 2015; Henderson et al., 2015; Henke et al., 2015; Li et al., 2015; Shi et al., 2015; Trimboli et al., 2015; Crescenzi et al., 2016; Haugen et al., 2016; Tavares et al., 2016]. This mutation leads to constitutive activation of the BRAF kinase and MAPK signaling pathway (Raf-MEK-ERK), being the most frequent molecular alteration in PTC (approximately 36-69%) [Cohen et al., 2003; Kimura et al., 2003; Namba et al., 2003; Nikiforova et al., 2003; Soares et al., 2003; Xu et al., 2003; Sobrinho-Simoes et al., 2008; Xing, 2010; Tavares et al., 2016]. It rarely coexists with RET or RAS mutations [Soares et al., 2003; Howell et al., 2013] appearing to be frequently associated, according to recent studies (Table 1), with TERT mutation (see below) [Vinagre et al., 2013]. In PTC, the BRAFV600E mutation displays a strong phenotype-genotype association, being almost exclusively detected in PTC with classic papillary or mixed growth (papillary/follicular pattern) both leading to a diagnosis of classic PTC [Tavares et al., 2016]. Its association with increased tumor aggressiveness and poor prognosis in PTC is still under debate. The BRAFV600E mutation is present in approximately half of the cases of classic PTC and in 40% of thyroid papillary microcarcinomas (mPTC), histotypes that have in general an excellent prognosis. In fact, most of the individuals with thyroid cancer with mutated BRAF evolve very well, turning the positive predictive value of this mutation to worse outcome per se very low. Recurrence is the most commonly described clinical and pathological feature associated with this mutation [Xing, 2010; Sancisi et al., 2012; Melo et al., 2015; Tavares et al., 2016]. In the majority of the series displaying associations between the BRAFV600E mutation and disease-specific mortality, the association, when present, is dependent on underlying clinical and pathological features [Fugazzola et al., 2006; Xing, 2007; Ito et al., 2009; Kwak et al., 2009; Lee et al., 2009; Handkiewicz-Junak et al., 2010; Kim TH et al., 2012; Kurtulmus et al., 2012; Ricarte-Filho et al., 2012; Sarne, 2012; Tufano et al., 2012; Choi et al., 2013; Xing et al., 2013, 2014, 2015; George et al., 2015; Melo et al., 2015; Shi et al., 2015]. Henke et al. [2015] corroborated these findings in a study involving 508 patients with PCT where the BRAF mutation was present in 67% of the cases. This mutation was predictive of capsular invasion, lymph node involvement, and classic PTC histology, but no relationship with recurrence-free survival or disease-specific survival was found, indicating that the BRAF mutation is not a predictor for recurrence and survival of PTC. In a previous study, Gouveia et al. [2013] showed that the BRAF mutation was not associated with tumor multicentricity, lymph vascular invasion, extra nodal extension, or advanced stage (III and IV) in a cohort of patients with PTC. In line with Henke et al. [2015], George et al. [2015] suggested that BRAF mutation is not a recurrence predictor in patients who develop a second recurrence after a first recurrence, but appears to be associated with recurrence after primary disease [Howell et al., 2011; Xing et al., 2015]. At variance with the above, Xing et al. [2013] verified that BRAF mutation was associated with disease-free survival in PTC. These discrepancies can be explained, at least in part, by differences in the follow-up times from the first surgery (median 33 months) in the study of Xing et al. [2013], compared to those of George et al. [2015] (9.3 years) and Henke et al. [2015] (8 years).
Summary of previous studies on TERT p and BRAF mutations in follicular-derived thyroid carcinoma and clinicopathologic associations
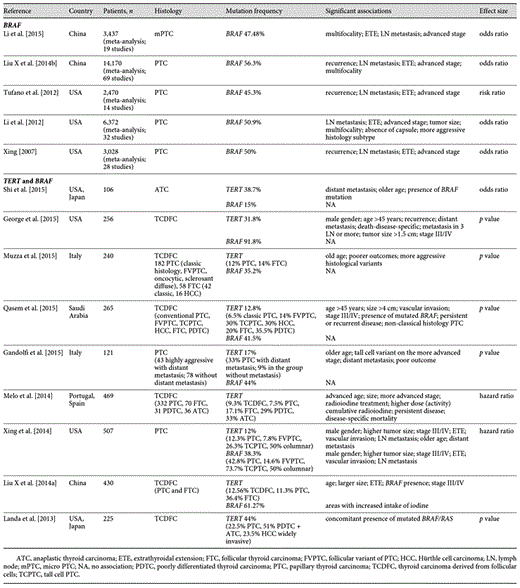
In a univariate analysis of 1,849 patients with PTC, the presence of the BRAFV600E mutation was associated with increased specific disease mortality, but this association loses its significance on multivariate analysis [Xing et al., 2013]. In a systematic review and meta-analysis of 14 publications that included 2,470 patients with PTC from 9 different countries, patients with tumors harboring the BRAFV600E mutation were associated with a higher risk of recurrence compared to patients without the mutation (24.9 vs. 12.6%, p < 0.00001, 95% CI 1.61-2.32), but with a positive predictive value of only 25% [Tufano et al., 2012]. In these meta-analysis studies [Tufano et al., 2012], the risk of recurrence in patients with BRAFV600E mutation ranged from 11 to 40% (median 26.5%), while the risk of recurrence in patients without this mutation was 2-35% (median 9.5%). Since the mutation is closely linked with histological features of aggressiveness (lymph node metastasis and extrathyroidal extensions), it is difficult to determine the proportion of risk that is attributable to the BRAF mutation per se or to the other clinicopathologic changes [Haugen et al., 2016]. Based on the aforementioned data, it appears that isolated BRAF mutation is not sufficient to substantially contribute to risk stratification in most PTC patients [Haugen et al., 2016].
In Table 1, we summarize several meta-analyses [Lee et al., 2007; Xing, 2007; Kim et al., 2012; Li et al., 2012; Tufano et al., 2012; Xing et al., 2014] that analyzed the BRAFV600E mutation in PTC (prevalence of mutation in the 45.3-56.3% range). In the majority of these meta-analyses, recurrence was associated with lymph node metastasis and extrathyroidal extension rather than with BRAF mutation (Table 1).
In addition to the above mutation (BRAFV600E), other BRAF mutations have been detected in PTC. Curiously, a genotype-phenotype correlation seems to exist also in those less frequent alterations: the BRAFK601E mutation is mainly seen in FVPTC, and the “in frame” deletion VK600-1E is detected in rare cases of the solid variant of PTC [Garcia-Rostan et al., 2003; Trovisco et al., 2005; Tavares et al., 2016]. An AKAP9-BRAF fusion rearrangement has been described in radiation-induced PTC [for a review, see Tavares et al., 2016].
TERT Mutations in TCDFC
A new genetic alteration, a mutation in the TERT promoter, TERT p, was recently described in TCDFC and other cancers [Horn et al., 2013; Huang et al., 2013; Rachakonda et al., 2013; Vinagre et al., 2013] and has been shown to be associated with increased aggressiveness and poor prognosis of the patients [Melo et al., 2015].
Telomerase is the enzyme involved in elongation of telomeres, complexes of nucleoproteins at the ends of the chromosomes that consist of several repetitions of the DNA sequence TTAGGG, whose main function is preservation of chromosome integrity and genomic stability [Cong et al., 1999; Cifuentes-Rojas and Shippen, 2012; Vinagre et al., 2014]. The telomere complex comprises different components, the most important of which are the telomerase reverse transcriptase catalytic subunit (TERT), the RNA component of telomerase (hTR), and dyskerin (DKC1 gene) [Cifuentes-Rojas and Shippen, 2012]. It has been shown that TERT is the only component required to restore the activity of the telomerase complex [Cong et al., 1999]. The TERT gene is located on chromosome 5 and includes 16 exons spanning a 35-kb region. The TERT promoter core includes 330 bp upstream of the transcription start (ATG) located in a GC-rich region containing binding sites for several transcriptions factors [Quiros et al., 2005].
For more than 20 years it has been known that high levels of telomerase can be detected in cancers, although, paradoxically, mutations in the coding region of the telomerase gene are uncommon in cancer [Kim et al., 1994; Vinagre et al., 2014].
Studies were published demonstrating mutations in TERT p in the central nervous system (43-51%), bladder (59-66%), hepatocellular (59%), thyroid (10%), and melanoma tissue (29-73%) [Killela et al., 2013; Nault et al., 2013; Pellegriti et al., 2013; Vinagre et al., 2013]. These mutations occur in 2 hot spot positions located -124 and -146 upstream from the ATG transcription start site. The mutation -124G>A, also called C228T, and the mutation -146G>A, also called C250T, are usually alternative and confer increased promoter activity of the TERT gene by creating a binding site (GGAA) for transcription factors of the ETS family within the TERT promoter region [Horn et al., 2013; Vinagre et al., 2014]. In accordance with the proposed mechanism of action, an increase of telomerase expression in tumors with mutated TERT p was shown [Vinagre et al., 2013].
Thyroid tissue rarely renews in adulthood. Consistent with this fact, telomerase activity in normal thyroid or benign thyroid tissues is almost absent (less than 7% of cases) [Capezzone et al., 2009; Soares et al., 2011; Vinagre et al., 2014]. Telomerase activity, however, is consistently reported in a specific population of normal thyroid cells (solid cell nests), that represent the remnants of the embryonic branchial body [Reis-Filho et al., 2003; Preto et al., 2004].
Thyroid carcinoma appears to present less frequently telomerase activation when compared to other types of human carcinomas, its expression being less common in WDTC (25-50%, depending on the series) than in PDTC or UTC (up to 100% of cases) [Soares et al., 2011].
Several studies have been published concerning TERT p mutations in thyroid carcinomas (Table 1). In the different series, the prevalence of TERT p mutation ranged from 7 to 50% [Landa et al., 2013; Liu X et al., 2013; Liu T et al., 2014; Melo et al., 2014; Xing et al., 2014], being present in 7-22% of PTC, in 14-22% of FTC, and with higher prevalence in patients with PDTC or UTC [Landa et al., 2013; Liu X et al., 2013, 2014a; Vinagre et al., 2013, 2014; Liu T et al., 2014; Melo et al., 2014; Xing et al., 2014].
In our hands [Melo et al., 2014], in a series of 469 TCDFC with a mean follow-up of 7.8 ± 5.8 years, we found TERT p gene mutation in 7.5% of PTC, 17.1% of FTC, 29% of PDTC, and 33.3% of UTC. TERT p mutation was significantly associated with distant metastasis in WDTC being considered an indicator of increased tumor aggressiveness, poor prognosis, and higher specific mortality due to the disease in WDTC in general, and in PTC in particular. Another important observation was that the coexistence of BRAF mutation in TERT-mutated PTC was not associated with increased aggressive clinicopathologic features or worse evolution. Also in this series [Melo et al., 2014], when analyzing all the factors associated with distant metastases in a logistic regression valuation model (gender, tumor size, vascular invasion, TERT mutation), vascular invasion became the only independent predictive factor for distant metastasis. However, vascular invasion is a post-surgical information, while all the others (gender, age, tumor size estimated by ultrasound, and TERT mutation in fine-needle aspiration material) can be obtained in the pre-surgical phase and can thus allow for better risk stratification before the initial treatment [Melo et al., 2014]. In this series, TERT p mutation was a predictor of lower disease-free survival and disease-specific mortality in WDTC [Melo et al., 2014]. Similar results were reported by others [Liu T et al., 2014; Xing et al., 2014; Gandolfi et al., 2015] that also showed the association of the TERT mutation with distant metastases in PTC (33 vs. 9% of TERT mutation in PTC cases with and without distant metastases, respectively) and its role in helping to predict increased aggressiveness of these tumors. In these studies, disease-specific mortality within the group with distant metastasis was not associated with TERT p mutation; this finding led the authors to suggest that this mutation probably plays a role in the acquisition of metastatic potential and that, once acquired, the TERT status does not continue to influence the evolution.
In contrast to the aforementioned results, in the study of Xing et al. [2014], TERT p mutation was more frequently found in PTC with BRAF mutation, and the combination of TERT and BRAF mutations was associated with an increased risk of structural disease [Xing et al., 2014]. In a large number of PTC, these authors observed that the coexistence of TERT p mutation -124C>T and BRAFV600E mutation was associated with worse outcome, worse clinicopathologic features, and increased recurrence when compared with tumors without such mutations. At variance with this, other groups [Melo et al., 2014; Muzza et al., 2015] showed that TERT mutation is strongly correlated with poor prognosis in patients with WDCT; in these 2 studies no additional effect of coexistent BRAF and/or RAS mutation was found. These conflicting results need confirmation from additional studies, but for the moment, we think there is enough evidence to claim that this molecular marker (TERT p mutation), alone or in combination with other genetic alterations, is useful for risk stratification of TCDFC and provides greater accuracy in risk appraisal of thyroid cancer in general [Landa et al., 2013; Liu et al., 2013; Melo et al., 2014; Haugen et al., 2016]. Qasem et al. [2015] showed the presence of TERT mutation in an ethnic group (Middle East patients) distinct from the others already studied and found this mutation in 12.8% of the patients. The mutation was present in 6.5% of classic PTC, 14% of FVPTC, 30% of tall cell variant of PTC, and in 38.5% of PDTC, reinforcing the results found in other series regarding the association of this mutation with more aggressive histology, BRAFV600E mutation, and persistence/recurrence of disease. In another study including 408 Chinese patients with thyroid cancer [Liu X et al., 2014a], it was shown that, unlike BRAF mutation, TERT p mutation is not associated with high iodine intake, but it is associated with older age, larger tumors, extrathyroidal extension, and advanced stages (III/IV) of PTC.
TP53 Mutations in TCDFC
Alterations in several tumor suppressor genes (encoding p53, Rb, p16, p21) have been associated with thyroid carcinogenesis [Pavelic et al., 2006]. Of these, the most studied and relevant is the TP53 gene, located on the short arm of chromosome 17 [Corapcioglu et al., 2006]. TP53 mutations are frequent in several human cancers being advanced as the most common genetic alteration in malignant cells [Morita et al., 2008]. The TP53 gene is activated following diverse stress stimuli, notably after DNA damage, and thus, this gene is frequently considered as the “guardian of the human genome” [Efeyan and Serrano, 2007].
The TP53 mutation in TCDFC is detected in exons 5-9, codon 273 being most commonly changed [Soares et al., 2011; Tavares et al., 2016]. In most cases, the TP53 mutation leads to expression of a dominant-negative mutant protein or, less commonly, to absence of protein expression [Levine and Oren, 2009; Lane and Levine, 2010; Tavares et al., 2016]. No TP53 mutation or abnormal p53 protein expression is seen in normal thyroid tissue, benign lesions, adenomatous goiter, and chronic thyroiditis [Tavares et al., 2016]. The prevalence of TP53 mutation/p53 overexpression in WDTC varies from 0 to 59% in the various studies on record [Nikiforov et al., 1996; Morita et al., 2008]. Nikiforova et al. [2013] have demonstrated that a small proportion of aggressive PTCs are associated with TP53 mutation and/or expression of p53 protein. p53 expression was described in the tall cell variant of PTC [Rüter et al., 1996], columnar/tall cell PTC [Putti and Bhuiya, 2000], squamous cell PTC [Kleer et al., 2000], some cases of the cribriform-morular variant of PTC [Jung et al., 2009], and the micro papillary/hobnail variant of PTC [Asioli et al., 2010b; Tavares et al., 2016] (see below). This mutation has been found in up to 25% of PDTC [Dobashi et al., 1993; Donghi et al., 1993; Pita et al., 2014; Tavares et al., 2016] and in over 60% of cases of UTC [Ito et al., 1992; Donghi et al., 1993; Soares et al., 2011; Pita et al., 2014; Tavares et al., 2016]. The expression of p53 is virtually limited to areas of thyroid tumors with undifferentiated components [Donghi et al., 1993; Ito et al., 1993; Pilotti et al., 1994; Quiros et al., 2005; Tavares et al., 2016].
TP53 mutation has been related to the transition of WDTC to UTC [Morita et al., 2008]. The loss of TP53 function could lead to dysregulation of the apoptotic pathways and genomic instability, and consequently promote the acquisition of additional oncogenic mutations. Several studies correlate the overexpression of p53 protein with the presence of TP53 gene mutation, justifying the absence of expression of the wild-type protein to its short half-life [Corapcioglu et al., 2006; Lim et al., 2007; Zafon et al., 2007; Morita et al., 2008]. According to these studies, the mutated form presents greater stability and a longer half-life as a direct consequence of the structural changes induced by the “missense” mutation, as highlighted in the study of Zafon et al. [2007], which attributed the immunohistochemical overexpression of p53 to gene mutation in up to 95% of the cases of PTC.
Quiros et al. [2005] evaluated the correlation between the BRAF mutation and TP53, suggesting that many UTC with PTC components are derived from BRAF-mutated PTC due to the acquisition of TP53 mutation. Zafon et al. [2007] analyzed the clinical significance of the expression of RET and p53 in PTC, and have shown that p53 expression was more prevalent in patients with RET/PTC rearrangements, significantly influencing the presence of extrathyroidal extension of the disease.
In a recent genomic analysis, TP53 mutations were identified in 3.5% (2/57) of PTC and in 11% (4/36) of FTC [Nikiforova et al., 2013]. The PTC patients with TP53 mutation in this series also showed mutation in BRAF (or BRAF and PIK3CA) and developed lung metastasis. All TP53-mutated FTC were oncocytic, and 3 out of 4 were widely invasive FTC [Nikiforova et al., 2013]. In the recent Cancer Genome Atlas (TCGA) study, 0.7% of the PTCs show TP53 mutations [Cancer Genome Atlas Research Network, 2014].
PIK3CA and AKT1 Mutations in TCDFC
Mutations in 2 other genes, PIK3CA and AKT1, are rarely identified in WDTC and are considered a late event in thyroid tumorigenesis, associated with tumor progression and dedifferentiation. In the TCGA study, which involved 400 patients with PTC, only 3 (0.8%) were shown to have an AKT1 mutation and 2 (0.5%) a PIK3CA mutation [Cancer Genome Atlas Research Network, 2014; Shrestha et al., 2015]. None of the aforementioned PTC had simultaneous occurrence of BRAF mutations with AKT1 and PIK3CA mutation, showing that multiple mutations are rare in PTC and, when they occur, BRAF and PIK3CA or AKT1 are found in more advanced tumors, less differentiated, and refractory to iodine [Ricarte-Filho et al., 2009; Nikiforova et al., 2013].
Molecular Alterations Associated with Aggressive Variants of Thyroid Carcinoma
Some variants of PTC are associated with high aggressiveness while others are associated with an indolent tumor behavior. The PTC variants with potentially more aggressive behavior are tall cell, columnar cell, hobnail, solid, and multinodular/diffuse [Ivanova et al., 2002] forms of FVPTC, and the diffuse sclerosing variant of PTC [Haugen et al., 2016].
The tall cell variant is characterized by predominance (>50%, according to some authors [Haugen et al., 2016]) of tall, oncocytic tumor cells whose height is at least 3 times their width. They are most frequently seen in old patients with a more advanced stage of neoplastic disease [Moreno Egea et al., 1993; Ostrowski and Merino, 1996; Leung et al., 2008] and show a higher recurrence rate and decreased disease-specific survival [Johnson et al., 1988; Moreno Egea et al., 1993]. The BRAFV600E mutation is found in about 80% of these tumors [Nikiforova et al., 2003; Xing, 2005; Haugen et al., 2016].
The columnar cell variant of PTC, characterized by predominance of columnar cells with pronounced nuclear stratification, has a higher risk of distant metastases and tumor-related mortality. The BRAFV600E mutation is found in about 33% of these tumors [Evans, 1986; Chen et al., 2011; Haugen et al., 2016].
The hobnail variant of PTC was recently described, being characterized by the predominance of cells with a hobnail appearance, with apically placed nuclei and bulging of the apical cell surface. This variant appears to have a higher risk of distant metastases (typically to lung) and an increased risk of tumor-related death [Motosugi et al., 2009; Asioli et al., 2010b; Haugen et al., 2016]. BRAF mutation and TP53 mutation/p53 expression are described in such tumors having TP53 mutations being implicated in the dedifferentiation process of the hobnail variant of PTC [Tavares et al., 2016].
The other variants of PTC that seem to behave more aggressively are the solid and multinodular/diffuse [Ivanova et al., 2002] form of FVPTC and the diffuse sclerosing variant. The latter carries a controversial prognosis. It tends to be seen in younger patients with good response to treatment. It is frequently characterized by diffuse involvement of the whole or part of the thyroid gland, higher rate of local and distant metastases (10-15%; predominantly lung), and lower disease-free survival than classic PTC. Nevertheless, the overall mortality appears to be low, with a disease-specific survival of approximately 93% (10 years of follow-up) which is not that different from classic PTC with similar neoplastic stages [Lam and Lo, 2006; Fukushima et al., 2009; Regalbuto et al., 2011; Haugen et al., 2016]. Pillai et al. [2015] found RET/PTC rearrangements in the diffuse sclerosing variant of PTC.
The PTC variant cited above, designated as “diffuse FVPTC”, was advanced in 1990 in order to identify a subset of PTC with a widely invasive follicular growth pattern, usually in a diffuse or multinodular fashion and involving 1 or both lobes of the thyroid, in contrast to the uninodular, well circumscribed, and frequently encapsulated appearance of common FVPTC [Ivanova et al., 2002]. This variant was described by the group of Sobrinho-Simões who showed the development of distant metastases - lungs and/or bones - with or without concurrent regional lymph node metastases in a series of 8 patients with diffuse FVPTC [Ivanova et al., 2002]. The clinicopathologic features of the multinodular/diffuse variant of FVPTC show unencapsulated pushing borders or absence of a clear-cut delineation between the tumor and the adjacent parenchyma, variable histologic features with a predominance of microfollicles and/or trabeculae, and frequent signs of vascular invasiveness, differing significantly from common PTC and FVPTC by its prevalence in younger patients and frequent multicentricity, extrathyroidal extension, nodal metastasis, and vascular invasion [Ivanova et al., 2002].
At the other end of the spectrum of the multinodular/diffuse variant of FVPTC, there is the noninvasive, encapsulated variant, E-FVPTC. As demonstrated in a study of Rosario et al. [2014], well-differentiated thyroid carcinomas that exhibit the following histological criteria are classified as noninvasive E-FVPTC: (1) tumor totally surrounded by a fibrous capsule; (2) follicular architecture; (3) unequivocal nuclear changes of PTC (multifocal or diffuse); and (4) absence of capsular or vascular invasion. Capsular invasion is defined as complete penetration of the entire thickness of the tumor capsule. Vascular invasion is defined as invasion of a vessel located in or outside the tumor capsule [Rosario et al., 2014]. In this study, the authors showed the excellent long-term evolution of patients with noninvasive E-FVPTC >1 cm when treated only by lobectomy (follow-up for 12-122 months, median 72 months, and no recurrence or elevation of Tg or TgAb was observed). This study suggested that in patients with noninvasive E-FVPTC lobectomy is sufficient, even in cases of tumors ≥1 cm and may spare some patients with thyroid tumors from the costs and risks of complementary surgery, 131I ablation, and TSH suppression. In accordance with this study, Nikiforov et al. [2016] showed no evidence of disease at the final follow-up (10-26 years, median 13 years) of all 109 patients with noninvasive E-FVPTC. The authors proposed a nomenclature revision for E-FVPTC with reclassification to “noninvasive follicular thyroid neoplasm with papillary-like nuclear features” (NIFTP), aiming to affect a large population of patients and to “result in a significant reduction in psychological and clinical consequence associated with the diagnosis of cancer” [Nikiforov et al., 2016].
Distinction between classical PTC and FVPTC morphology has been validated at the molecular level by the Cancer Genome Atlas (TCGA) Research Network. The PTCs were classified into BRAFV600E-like and RAS-like tumors with distinct genomic features, the classical PTCs generally being placed into the BRAFV600E-like category, whereas PTCs with follicular architecture and nuclear atypia (multifocal or diffuse) - FVPTC - were placed into the RAS-like category. Based on the expression of a number of thyroid genes, the authors determined the thyroid differentiation scores (TDS), which support evidence that FVPTC has a gene expression profile similar to the normal thyroid, and classical PTCs (BRAF-like) display less evident thyroid differentiation with lower expression levels of TDS genes [Cancer Genome Atlas Research Network, 2014; Asa et al., 2015]. The RAS-like group seemed to represent a single entity; by contrast, the BRAFV600E-like group comprised WDTC and a category with much less differentiation and, therefore, a heterogeneous group of thyroid cancer [Cancer Genome Atlas Research Network, 2014; Asa et al., 2015].
Solid PTC appears to be more frequently associated with distant metastases (15% of cases) and with a higher mortality rate (10-12%) [Nikiforov et al., 2001; Haugen et al., 2016], except among children and adolescents in the context of post-Chernobyl PTC, that frequently displays a solid pattern of growth and very low mortality (<1%) during the first 10 years of follow-up [Cardis et al., 2006; Nikiforov, 2006; Haugen et al., 2016]. It is important to distinguish this variant from PDTC, which has a worse outcome, with a 10-year survival of about 50%. Both have the same growth patterns - insular, solid, and/or trabecular - and the distinction is based primarily on the preservation of nuclear features of PTC, lack of necrosis, and low mitotic activity in the solid variant of PTC, following the Turin diagnostic criteria [Volante et al., 2007; Asioli et al., 2010a]. According to other authors, the diagnostic boundary between PDTC and the trabecular or solid variant of PTC - both with island growth or solid or trabecular growth - is based exclusively on the maintenance of nuclear characteristics in the latter group, despite the presence of foci of necrosis and/or high mitotic activity [Sobrinho-Simoes et al., 2002; Garcia-Rostan and Sobrinho-Simoes, 2011; Tavares et al., 2016].
Besides the already referred genetic alterations in PDTC (in TERT p, BRAF, NRAS, TP53), other genetic alterations are relevant in PDTC.
Unlike WDTC, where NRAS codon 61 mutation is most commonly observed, KRAS and HRAS mutations in codons 12, 13, and 61 occur in PDTC and UTC [Sobrinho-Simoes et al., 2008; Nikiforov and Nikiforova, 2011; Soares et al., 2011; Tavares et al., 2016]. In PDCT, PIK3CA mutations are found in 5-14% of cases, PTEN mutations in about 20% of cases, and AKT1 mutations in 5-10% of cases [Garcia-Rostan et al., 2005; Nikiforov and Nikiforova, 2011; Pita et al., 2014; Eloy et al., 2015]. Ricarte-Filho et al. [2009] showed an association between BRAF-mutated PDTC and radioiodine refractory status.
As noted above, in UTC, inactivation of the TP53 gene appears to have a determinant role, being a late event in the tumorigenic process and occurring simultaneously with a huge increase in cell proliferation [Sobrinho-Simoes et al., 2008; Ricarte-Filho et al., 2009; Eloy et al., 2015]. BRAF mutation is also seen in up to 40% of cases of UTC [Kimura et al., 2003; Nikiforova et al., 2003; Soares et al., 2004; Ricarte-Filho et al., 2009; Eloy et al., 2015]. UTC presents the highest rates for mutations in TERT p (up to 50%) [Asioli et al., 2010a; Hannallah et al., 2013; Pita et al., 2014; Eloy et al., 2015]. In a study by Shi et al. [2015], the authors analyzed the association of TERT p mutation and clinical and pathological features of 106 patients with UTC. They verified that TERT p mutations in UTC were not associated with tumor characteristics such as size, extrathyroidal invasion, and lymph node metastasis that are probably intrinsic characteristics of UTC. Remarkably, TERT p mutations were associated with advanced age and distant metastasis in UTC, demonstrating also the prognostic role of this mutation in these highly aggressive cancers [Shi et al., 2015].
Our group reported that the LRP1B expression level in UTC was significantly lower than in WDTC, and that such reduced expression was due to mutation and genomic loss of the LRP1B gene [Prazeres et al., 2011]. UTC showed frequent methylation of the promoter region of the gene, leading to loss of LRP1B expression in more than 80% of UTC [Beroukhim et al., 2010; Prazeres et al., 2011; Eloy et al., 2015]. The role of LRP1B in advanced thyroid cancer deserves further investigation.
Isolated and Associated Mutations and Their Role in Predicting Thyroid Cancer Outcomes
The majority of the studies to date indicate that TERT p mutation can serve as a marker of more aggressive disease in all the subtypes of thyroid carcinomas, being the best current marker of poor prognosis, due to its association with distant metastases and increased disease-specific mortality [Melo et al., 2014].
The possible prognostic effect of the coexistence of BRAF and TERT p mutations has been suggested by some studies [Sancisi et al., 2012; Liu X et al., 2014a; Xing et al., 2014; Shi et al., 2015] but not by others [Landa et al., 2013; Melo et al., 2015; Muzza et al., 2015]. This controversy requires larger multicenter studies to definitely clarify whether the coexistence of BRAF and TERT p mutations in the same tumor has a prognostic value distinct from the presence of TERT p mutation alone. George et al. [2015] evaluated the effects of BRAFV600E mutation in association with TERT p mutation on recurrence and survival of high-risk patients, carriers of persistent or recurrent thyroid carcinomas and showed that only TERT p mutation, but not BRAF mutation, was associated with decreased survival (distant metastasis to lung or bone was significantly higher in patients with TERT p mutation). The association of TERT and RAS mutation was also reported to have an impact on the patient's prognosis [Landa et al., 2013]. Although less studied than other associations, there are also discordant results in the literature discussing whether or not there is a possible additional effect of RAS mutation in a TERT p-mutated WDTC with respect to the persistence of the disease [Landa et al., 2013; Song et al., 2016].
With subsequent confirmation of these data in larger series, and/or series with more aggressive tumors, it is likely that in the future TERT p mutation may be useful in individualizing treatments such as type of surgery and radioiodine therapy, as well as follow-up. Larger multicenter studies are needed to assess the prognostic impact of the coexistence of BRAF mutation and/or RAS and/or RET/PTC and/or TP53 with mutated TERT p in TCDFC.
Concluding Remarks
Most WDTC have indolent behavior, and the early identification of high-risk patients who may exhibit a more aggressive course of the disease is important in order to try to find a way of preventing unfavorable developments [Shrestha et al., 2015].
Several studies have shown the feasibility of mutational analysis in fine needle biopsy aspirates [Xing et al., 2014; Shrestha et al., 2015; Crescenzi et al., 2016] allowing preoperatively to evaluate the mutational tumor profile. The benefits of this approach are evident, for example, in undetermined cytology specimens in which the identification of a BRAFV600E mutation is almost 100% diagnostic of PTC. Similarly, TERT p mutation defines the diagnosis of malignancy in a thyroid nodule and also preoperatively identifies a TCDFC with higher aggressive potential, enabling better risk stratification [Liu X et al., 2014b]. Following the same reasoning, some authors suggest that a more accurate prediction of thyroid cancer outcome is possibly based on a more extensive genetic analysis, since, as discussed above, some data suggest a more aggressive clinical course in those patients harboring tumors with combination of other mutations such as TERT p and BRAFV600E or TERT p and RAS. The same is true concerning the identification of other mutations that are only relatively frequent in advanced tumors (e.g., TP53, PIK3CA, AKT1, or TERT p).
In the recent revision of the American Thyroid Association guidelines, the genetic profile was introduced to help stratifying the risk among patients classified as low risk. This was mainly based on a series of patients with PTC <4 cm, N0M0 [Elisei et al., 2012]. The overall risk of recurrence as structural disease at 5 years follow-up was 3% (8% in mutated BRAF and 1% in wild-type BRAF). Yet, in a multivariate analysis, the only significant clinicopathologic predictor of persistent disease after 5 years of follow-up was the presence of the BRAFV600E mutation [Elisei et al., 2012].
The impact of BRAF mutation in patients with mPTC is also disputable. Although this mutation is found in about 30-67% of these patients, the overall recurrence rate is very low ranging between 1 and 6% [Nikiforova et al., 2003; Sedliarou et al., 2004; Kim et al., 2006; Lim et al., 2007; Xing, 2007; Lin et al., 2010; Zheng et al., 2013; Haugen et al., 2016]. This is not true for multifocal, not incidental mPTC with extrathyroidal extension and BRAFV600E mutation in which the recurrence rate rises to 20%, leading these patients, which comprise about 10% of cases of mPTC, to be staged as intermediate risk [Haugen et al., 2016].
In summary, we show in this review that, at present, TERT p mutation is consolidating itself as a strong independent prognostic predictor in thyroid cancer. A better understanding of this mutation, together with additional ones like BRAF, RAS, or TP53, and their identification in fine-needle aspiration biopsies are likely to allow an early refinement of the stratification risk in patients with WDTC in the future. It is worth noting, however, that, as with any categorical staging system, the risk within the individual risk categories (low, intermediate, and high) can vary depending on the specific clinicopathologic features of individual patients and the specific biological behavior of the tumor [Haugen et al., 2016]. Finally, besides the diagnostic or prognostic significance of the above-mentioned mutations, their molecular pathways and epigenetic alterations will likely be targets for new therapies [Tavares et al., 2016].
Acknowledgement
This study was supported by a CNPq PhD Scholarship (“National Counsel of Technological and Scientific Development”, Brazil), Science Without Borders, and CAPES Foundation Ministry of Education of Brazil, Brasília - DF 70.040-020, Brazil, Process No. BEX6726/15-1 to G.P. This work was financed by FEDER, Fundo Europeu de Desenvolvimento Regional, funds through the COMPETE 2020 - Operational Programme for Competitiveness and Internationalisation (POCI), Portugal 2020, and by Portuguese funds through FCT - Fundação para a Ciência e a Tecnologia/Ministério da Ciência, Tecnologia e Inovação in the framework of the project “Institute for Research and Innovation in Health Sciences” (POCI-01-0145-FEDER-007274). Further funding from the project “Advancing cancer research: from basic knowledgement to application”; NORTE-01-0145-FEDER-000029; “Projetos Estruturados de I&D&I”, funded by Norte 2020 - Programa Operacional Regional do Norte.
Disclosure Statement
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
