

Abstract
Background/Aims: Ischemic stroke is a leading cause of long-term disability. To date, there is no effective treatment for stroke. Previous studies have shown that Ginkgo biloba extract has protective effects against neurodegenerative disorders. In this present study, we sought to test the potential protective role of an active component of Ginkgo biloba extract, bilobalide, in a rat model of middle cerebral artery occlusion (MCAO). Methods: A rat model of MCAO was used to test the potential protective effects of Bilobalide B on stroke protection. TTC staining was performed to evaluate infarct size of the brains. Neurological deficit score was measured to reveal the effects of the treatments on animal behavior and cognition. Immunohistochemical staining and transmission electronic microscope analysis were performed to measure the cellular responses to drug treatment. Western blotting and ELISA were performed. The expression of Cleaved- Casepase 3, Beclin-1, p62 and LC3I/II were quantified, and the Phosphorylation of eNOS and Akt were evaluated. The ratio of Bcl-2/ Bax was determined to reveal the molecular pathways that are involved in the drug treatment. Results: We found that intraperitoneal delivery of various Bilobalide doses during ischemia can protect against brain injury, as evidenced by reduced infarct size and improved neurological scores after surgery. Histochemical analysis revealed that treatment with bilobalide can significantly reduce apoptosis, autophagy, and promote angiogeneis following ischemia/reperfusion injury to the brain. The performence of increased phosphorylation of eNOS and Akt suggested that bilobalide can activate Akt prosurvival and eNOS pathways to promote cell survival and angiogenesis, respectively. Conclusions: Our results suggested that bilobalide benefits stroke symptoms by reducing cell death pathways and promoting angiogenesis. As such, bilobalide may be a potential agent for improving self-repair after ischemic stroke.
Introduction
Ischemic stroke is a leading cause of long-term disability. Every year, over 800, 000 Americans suffer from this life-threatening disease, with 40% of fatal cases [1]. Occlusion of cerebral arteries, such as the middle cerebral artery, causes ischemic stroke. An ischemic stroke leads to a number of neurotoxic events, including oxidative stress, inflammation, and calcium overload, which finally leads to the death of neurons, and other cell types within the brain [2, 3]. Although previous studies have identified some neuroprotective agents, such as cinnamophilin, Epigallocatechin gallate (EGCG), and oxyresveratrol, their beneficial effects are still very limited. Thus, novel neuroprotective agents are highly desired for treatment of this disease.
The constituents of the Ginkgo biloba extract EGb761 are widely used to treat various disorders, including cerebralinsufficiency Alzheimer's disease, dementia, and cerebral ischemic disease [4-6]. Bilobalide, a sesquiterpene lactone that constitutes 2.9–3.2% of the Ginkgo extract EGb761, has been shown to have a wide variety of therapeutic applications on neurodegenerative disease and dementia [7]. Other studies found that bilobalide may improve the outcome of ischemic dementia by promoting the mitochondrial respiratory chain function [8, 9] and down-regulaing nitric oxide [10]. However, the specific molecular mechanism underlying bilobalide mediated dementia protection needs to be further invesitgated.
Apoptosis and necrosis are important pathways of neuronal death induced by cerebral ischemia, but, in recent years, a third type of cell death, autophagy, has been discovered and studied [11, 12]. Autophagy occurs at a basal level in the absence of cerebral ischemia, fulfilling a cytoprotective role that promotes cell survival under conditions of nutrient deprivation. The protective role of autophagy during reperfusion may be attributable to mitophagy-related mitochondrial clearance and inhibition of downstream apoptosis [13]. However, under certain conditions, autophagy may promote cell death, and inhibition of which has been shown to reduce neurodegenerative damage after focal cerebral ischemia [14, 15]. As such, inhibition of autophagy may represent a novel strategy to protect against ischemic brain injury [16].
We have previously reported that autophagy downregulation improved recovery following stroke by promoting neurogenesis [17]. Wei Naokang (WNK), the active compounds extracted from ginkgo, ginseng, and saffron, has been demonstrated to have therapeutic effects on vascular dementia. Based on these beneficial effects, a Phase III clinical trials in Australia have begun. We found that WNK, and its active compound bilobalide, inhibited neuron autophagy and improved neurogenesis in ischemic peripheral areas. This suggested that autophagy might be a intervention target by which bilobalide promotes self-repair of ischemic areas [18].
Based on these findings, we hypothesized that bilobalide may be utlized in stroke treatment via inhibing autophagy and promoting neurogenesis or angiogenesis. In this present study, bilobalide was used in a rat model of middle cerebral artery occlusion (MCAO). We found that intraperitoneal (IP) delivery of various doses of bilobalide during ischemia can protect against brain injury, as evidenced by reduced infarct size and improved neurological scores after surgery. Histochemical analysis revealed that treatment with bilobalide can significantly reduce apoptosis, autophagy, and promote angiogeneis following ischemia/ reperfusion (I/R) injury to the brain. Biochemical assays meant to determine the molecular mechanisms underlying bilobalide mediated neuroprotective effects suggested that it can activate Akt prosurvival and eNOS pathways to promote cell survival and angiogenesis, respectively. Co-treatment of animals with a combination of bilobalide and inhibitors of Akt and/or eNOS pathways suppported our biochemical observations. Treatment with Akt and eNOS inhibitors partially abolished the beneficial effects of bilobalide, indicating that it does protect against neuronal injury through activation of these signaling pathways. Taken together, our results suggest that bilobalide benefits post-ischemia stroke symptoms by promoting angiogenesis and reducing both apoptosis and autophagy. Thus, bilobalide may be a potential agent for improving self-repair following ischemic stroke.
Materials and Methods
Animals
Adult male Sprague–Dawley rats (220–230 g) , provided by the Animal Facility of Peking University Health Science Center(Beijing, China), were housed in the laboratory animal room and maintained at 25±1°C with 65±5% humidity on a 12 hour light/dark cycle (lights on from 07: 30 to 19: 30) for at least 1 week before experiments. Animals were given food and water ad libitum. All experimental protocols described in this study were approved by the Ethics Review Committee for Animal Experimentation of Xiyuan Hospital, China Academy of Chinese Medical Sciences. All methods in this study were performed in accordance with the relevant guidelines.
Transient MCAO
Rats were subjected to transient focal cerebral ischemia induced by right MCAO as previously described [19, 20]_ENREF_19, with some modifications. In brief, rats were anesthetized with 10% chloral hydrate (360 mg/kg, i.p.), and arterial blood samples were collected via femoral catheter to measure PO2, PCO2 and pH with an AVL 998 Blood Gas Analyzer (Roche Co., Basel, Switzerland). Rectal temperature was maintained at 37±0.5°C during MCAO using a temperature-regulated heating lamp. A fiber-optic probe was attached to the parietal bone overlying the middle cerebral artery, 5 mm posterior and 5 mm lateral to bregma, and connected to a laser Doppler flowmeter (Perifluxsystem 5000, Sweden) for continuous monitoring of cerebral blood flow (CBF). A 4-0 nylon monofilament suture with a heat-blunted tip was introduced into the internal carotid artery through the stump of the external carotid artery, then gently advanced for a distance of 18 mm from the common carotid artery bifurcation to block the origin of the middle cerebral artery for 90 min, and then withdrawn to allow reperfusion. Only animals that exhibited > 85% CBF reduction during right MCAO and > 80% CBF recovery after 10 min of reperfusion were included in the study. After the wound had been closed, animals were allowed to recover from anesthesia before they were returned to their home cages.
Drug administration
Bilobalide B was isolated by chromatography of ginkgo extract (Chemistry Lab, Institute of Basic Medical Sciences, Xiyuan Hospital, Beijing). Stock solutions were prepared from 100 mM DMSO to yield concentrations of 5, 10, or 20 mg bilobalide B/kg body weight during treatment. NG-nitro-arginine methyl ester (L-NAME), an NOS inhibitor, was purchased from Cell Signaling Technology (Danvers, MA, USA), and LY294002 (LY), a PI3 kinase inhibitor, was purchased from Sigma-Aldrich (St. Louis, MO, USA). Unless otherwise indicated, all other chemicals used were of analytical grade and were purchased from Sigma-Aldrich.
Protocol one: To determine whether bilobalide B inhibits autophagy and apoptosis in the brain 24 hours after MCAO/reperfusion by activating the Akt and eNOS pathways, rats were randomly divided into different groups (n=12 for each group). These groups were sham control, I/R, I/R plus bilobalide B (5, 10, or 20 mg/kg), and I/R plus bilobalide B (10 mg/kg) after pretreatment with L-NAME (5 µl of 5 mM solution per animal) or LY294002 (5 µl of 100 µM solution per animal), stereotaxically delivered into the ipsilateral lateral ventricle 30 min before MCAO. Bilobalide B was delivered by way of duodenal administration between ischemia and reperfusion (normal saline was administered for sham and I/R groups).
Protocol two: In brief, rats were randomly divided into 6 groups (n=10 for each group), sham control, I/R, I/R plus bilobalide B (10 mg/kg), and I/R plus bilobalide B (10 mg/kg) after pretreatment with L-NAME (5 µl of 5mM solution per animal) and/or LY294002 (5 µl of 100 µM solution per animal), stereotaxically delivered into the ipsilateral lateral ventricle 30 min before and 48h after MCAO. Additionally, rats were treated with bilobalide B or a vehicle control (normal saline for sham and I/R groups) once daily, 24h, 48h, and 72h after MCAO. Duodenal administration of bilobalide B occured between ischemia and reperfusion (normal saline was administered for sham and I/R groups).
Measurement of Infarct Volume
At 24 hours or 7 days post reperfusion, rats were deeply anesthetized with 3.5% chloral hydrate and then decapitated. Whole brains were rapidly removed, and 2 mm coronal sections were stained with standard 2% 2, 3,5-triphenyltetrazolium chloride (TTC, Sigma-Aldrich) for 10 minutes at 37°C followed by overnight immersion in 4% formalin. Infarct volume, calculated by integration of infarct area on each brain section along the rostral-caudal axis and expressed as a percentage of whole-brain volume, was measured by an image processing and analysis system (1.25 × objective, Q570IW, Leica, Germany) [21].
Assessment of neurological deficit score
Neurological deficit scores were determined at 24 hours and 7 days after MCAO as previously described [22]. In brief, scores were determined as follows: no neurological deficit (value=0); left forelimb flexion when suspended by the tail or failure to fully extend right forepaw (value=1); left shoulder adduction when suspended by the tail (value=2); reduced resistance to lateral push toward the left side (value=3); spontaneous movement in all directions with circling to the left exhibited only if pulled by tail (value=4); circle or walk spontaneously only to the left (value=5); walk only when stimulated (value=6); no response to stimulation (value=7); and stroke-related death (value=8).
Transmission Electron Microscopy (TEM)
TEM was used to evaluate ultrastructural changes to brain sections. Cerebral fragments were fixed with 2.5% glutaraldehyde solution overnight at 4°C,were washed with PBS, and then fixed with 1% osmic acid for 2 h. Tissues were embedded in an Epon Araldite mixture. Ultra-thin sections were cut and stained with uranyl acetate and lead citrate. The samples were observed under a 1230 type transmission electron microscope (Electron Co., Japan) and photographed.
Immunofluorescence confocal imaging
To label angiogenesis, bromodeoxyuridine (BrdU, 100 mg/kg, Sigma) was administered (i.p) daily for 4 consecutive days starting at the 3 day after stroke. Rats were sacrificed 7 days after MCAO with an overdose of 3.5% chloral hydrate and were transcardially perfused with 0.9% saline solution followed by 4% ice-cold phosphate-buffered paraformaldehyde (PFA). Brains were removed, postfixed overnight, and equilibrated in phosphatebuffered 30% sucrose. Coronal sections at 1.0 to 0.2 mm from bregma were used for immunohistochemical staining and were cut on a freezing microtome (Leica CM3000, Germany) at a thickness of 25 μm. Frozen sections were double-stained for immunofluorescence and cell phenotypes were identified by colocalization of phenotypic markers with BrdU, as previously described, using these primary antibodies (and their concentrations): BrdU Mouse mAb (1: 100; cell signal); rabbit anti-CD31 antibody (1: 50; Cell Signaling); Secondary antibodies: anti-mouse and rabbit IgG-fluorescein isothiocyanate (FITC) and IgG-Cy3 (1: 200; Chemicon). Confocal images were taken with a Zeiss LSM-510 microscope.
Western Blotting
Rats were sacrificed 6h, 24h or 7d after reperfusion with an overdose of 3.5% chloral hydrate. Cortical sections 1.0 to 2.0 mm from infarcts were used for western blot analysis. Rat cerebral homogenates were collected and centrifuged for 4 min at 3000×g and the resulting supernatants spun at 10, 000×g for 30 min. Supernatants were diluted to a protein concentration of 0.5 mg/ml for measurement of Bcl-2, Bax, Beclin 1, and LC3 expression and for the phosphorylation of Akt and eNOs. Protein content was determined with BSA as a standard according to Bradford [23]. Protein samples (20 μg/lane) were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and were transferred to PVDF membranes (Millipore, Billerica, MA) through electroblotting. Blots were stained with rabbit polyclonal anti-autophagy APG8a (MAP1LC3A) antibody (Abgent), rabbit polyclonal anti-Beclin1 antibody (Cell Signaling), Rabbit mAb phosphorylated-Akt (Ser473) and total Akt antibody (Cell Signaling), Anti-phospho-eNOS (pSer1176) and total eNOS antibody (Sigma-Aldrich). Blots were then developed by enhanced chemiluminescence using SuperSignal West Femto maximum sensitivity substrate (Pierce, Rockford, IL, USA). Bio-Rad Image LabTMVersion 3.0 software was used to calculate the numerical value of every blot. The mean densitometric×area values were depicted as bar graphs. All western blots reported in this study were performed three times, and the results were reproducible
VEGF and NO ELISA assay
The tissue homogenates containing VEGF and NO were centrifuged (1000×g, 10 min, 4 °C) and the concentrations of VEGF in the supernatants were measured with ELISA kits, according to the manufacturers’ directions(R&D systems). The concentrations of NO were measured with the total Nitric Oxide assay kit (Beyotime Biotechology). Sample and standard dilutions were made with the experimental media, and the results are expressed as the means±SD.
Data Quantification and Statistical Analysis
All data are presented as mean±SD. Statistical significance was analyzed using a one-way analysis of variance (ANOVA) followed by Tukey’s test for multiple comparisons. A nonparametric test was used for comparison of band density values between groups. P< 0.05 was considered statistically significant.
Results
Bilobalide protects against infarct and associated neurological deficits following stroke
Rats were sacrificed 24 hours and 7 days following transient focal cerebral ischemia induced by right MCAO, their brains were quickly isolated, and coronal sections were stained with TTC in order to determine infarct volume as a percentage of total brain volume. It was found that bilobalide treatment significantly reduced infarct volume, as compared to saline treatment at both time points. Examination of serial brain sections showed that bilobalide treatment (10 and 20 mg/kg) significantly decreased the infarct volume compared with the I/R group 24h after focal ischemia/reperfusion. The neurological deficit scores decreased in the I/R group, and significant differences were found at 24 h after reperfusion compared with the bilobalide (10 and 20 mg/kg) treatment group (Fig. 1). These findings indicate that bilobalide protects against ischemic brain injury and the dose of 10 mg/kg was used in the next study. Additional studies indicated that Rd (10 mg/kg) significantly increased angiogenesis 7 d after focal ischemia/reperfusion (Fig. 2). At 24 hours, bilobalide (10 mg/kg) treatment reduced infarct volume from 39% to 31%, and, at 7 days, bilobalide (10 mg/kg) treatment reduced infarct volume from 28% to 13% (Fig. 2A). In order to determine whether the reduced infarct volume was associated with improved neurological performance, rats were evaluated for deficits 24 hours and 7 days following I/R injury. Each rat was scored 0-8, with 0 being the best score (no neurological deficit) and 8 being the worst (stroke related death). Treatment with bilobalide (10 mg/ kg) increased performance at both time points, with average deficit scores being reduced from 7.1 to 4.1 at 24 hours, and from 6.8 to 3.8 at 7 days (Fig. 2B). In order to explore the beneficial effects of bilobalide treatment, we performed confocal analysis to probe samples with CD31 (an angiogenesis biomarker) and BrdU (a cell proliferation biomarker). As shown in Fig. 2C, confocal microscopic analysis of CD31 and BrdU stained tissues revealed that bilobalide treatment significantly increases the angiogenesis and cell proliferation associated with recovery from I/R injury.

Infarct volume and neurological scores. (A) Representative TTC staining of the cerebral infarct in comparable sections of rat brain, 24h after ischemia. Scale bar=0.5cm (B) Quantification of infarct volume. (C) Neurological scores. Mean±SD. n=12. **P< 0.01 vs I/R group.
Infarct volume and neurological scores. (A) Representative TTC staining of the cerebral infarct in comparable sections of rat brain, 24h after ischemia. Scale bar=0.5cm (B) Quantification of infarct volume. (C) Neurological scores. Mean±SD. n=12. **P< 0.01 vs I/R group.

Bilobalide protects against I/R injury to rat brain. (A) Representative images of TTC staining of brain slides show that treatment with bilobalide reduced infarct area 24 hours and 7 days after I/R injury. Saline treatment was used as a negative control. Infarct area, as determined by multimedia color pathological image analysis (MPIAS-500, Beijing Konghai Company, China) (right panel), was significantly reduced in bilobalide (10mg/kg) treated rats as compared to those that were saline treated. Scale bar=0.5cm (B) Bilobalide (10mg/kg) treatment also improvedneurological deficit scores. (C) 7 days after I/R injury, angiogenesis and cell proliferation were quantified by immunostaining of CD31 and BrdU. Treatment with bilobalide (10mg/kg) significantly enhanced angiogenesis and cell proliferation as compared to saline treatment. Data are presented as mean±SD. n=12 rats per group at 24 hour time point; n=10 rats per group at 7 day time point. ΔΔ, ## or ** : p< 0.01.
Bilobalide protects against I/R injury to rat brain. (A) Representative images of TTC staining of brain slides show that treatment with bilobalide reduced infarct area 24 hours and 7 days after I/R injury. Saline treatment was used as a negative control. Infarct area, as determined by multimedia color pathological image analysis (MPIAS-500, Beijing Konghai Company, China) (right panel), was significantly reduced in bilobalide (10mg/kg) treated rats as compared to those that were saline treated. Scale bar=0.5cm (B) Bilobalide (10mg/kg) treatment also improvedneurological deficit scores. (C) 7 days after I/R injury, angiogenesis and cell proliferation were quantified by immunostaining of CD31 and BrdU. Treatment with bilobalide (10mg/kg) significantly enhanced angiogenesis and cell proliferation as compared to saline treatment. Data are presented as mean±SD. n=12 rats per group at 24 hour time point; n=10 rats per group at 7 day time point. ΔΔ, ## or ** : p< 0.01.
Bilobalide reduces apoptosis and autophagy following I/R
In order to determine whether these protective effects were at all mediated by changes in the amount of apoptosis following stroke, brain slides from sham control rats, I/R rats, and I/R rats treated with bilobalide (10 mg/kg) were stained with antibody targeting cleaved caspase-3, an apoptosis indicator. The number of cells positive for cleaved caspase-3 was significantly reduced in the bilobalide (10 mg/kg) treated group as compared to the saline control, indicating that bilobalide is able to reduce stroke induced apoptosis (Fig. 3).

Bilobalide treatment inhibits apoptosis induced by I/R injury to the brain. Tissue slides were stained with antibody against cleaved caspase-3 as an indicator for apoptosis. Bilobalide (10mg/kg) treatment significantly reduced caspase-3 positive cells (quantified by ImageJ software in lower panel) 24 hours and 7 days after I/R injury. Data are presented as mean±SD. n=12 rats per group at 24 hour time point; n=10 rats per group at 7 day time point. ## or ** : p< 0.01.
Bilobalide treatment inhibits apoptosis induced by I/R injury to the brain. Tissue slides were stained with antibody against cleaved caspase-3 as an indicator for apoptosis. Bilobalide (10mg/kg) treatment significantly reduced caspase-3 positive cells (quantified by ImageJ software in lower panel) 24 hours and 7 days after I/R injury. Data are presented as mean±SD. n=12 rats per group at 24 hour time point; n=10 rats per group at 7 day time point. ## or ** : p< 0.01.
It has been shown that autophagy also contributes pathologies associated with I/R injury to the brain. In order to determine whether bilobalide treatment can regulate autophagic pathways, transmission electron microscopy was performed. The results revealed that following I/R, autophagy levels were reduced in bilobalide (10 mg/kg) treated rats as compared to those that were saline treated. That is, bilobalide (10 mg/kg) significantly reduced the appearance of autophagic vacuoles following I/R (Fig. 4A). Furthermore, western blot analysis of cerebral homogenates showed that autophagy-related markers, Beclin-1 and LC3-II/LC3-1 ratio, were significantly reduced in bilobalide (10 mg/kg) treated rats. On the contrary, p65 expression increased in bilobalide (10 mg/kg) treated rats (Fig. 4B). Immunostaining showed distinct punctation of LC3-II around brain endothelial cells following I/R injury. However, the amount of this punctation was reduced to almost non-injured levels in cells from bilobalide (10 mg/kg) treated rats (Fig. 4C).

Bilobalide treatment suppresses autophagy following I/R injury to the brain. (A) Representative electron microscope images show that I/R injury induced formation of autophagic vacuoles (identified by arrows), but that treatment with bilobalide (10mg/kg) could inhibit their formation. Summary of upper panels was showed in lower panel. (B) Western blot analysis showed that treatment with bilobalide (10mg/kg) significantly suppressed Beclin-1 and the LC3-II/LC3-I ratio, two key autophagy related markers, following I/R injury. (C) LC3 foci formation (foci showed in red) was visualized using a Zeiss LSM-510 microscope (upper panels) and quantified by Image-Pro Plus (lower panel). Treatment with bilobalide (10mg/kg) reduced LC3 foci formation induced by I/R injury. Data are presented as mean±SD. n=12 rats per group at 24 hour time point; n=10 rats per group at 7 day time point. ΔΔ, ## or **: p< 0.01; Δ, # or * : p< 0.05.
Bilobalide treatment suppresses autophagy following I/R injury to the brain. (A) Representative electron microscope images show that I/R injury induced formation of autophagic vacuoles (identified by arrows), but that treatment with bilobalide (10mg/kg) could inhibit their formation. Summary of upper panels was showed in lower panel. (B) Western blot analysis showed that treatment with bilobalide (10mg/kg) significantly suppressed Beclin-1 and the LC3-II/LC3-I ratio, two key autophagy related markers, following I/R injury. (C) LC3 foci formation (foci showed in red) was visualized using a Zeiss LSM-510 microscope (upper panels) and quantified by Image-Pro Plus (lower panel). Treatment with bilobalide (10mg/kg) reduced LC3 foci formation induced by I/R injury. Data are presented as mean±SD. n=12 rats per group at 24 hour time point; n=10 rats per group at 7 day time point. ΔΔ, ## or **: p< 0.01; Δ, # or * : p< 0.05.
Bilobalide reduces I/R damage in an Akt and eNOS dependent manner
Nitric oxide (NO) production and levels of vascular endothelial growth factor (VEGF) have been demonstrated to play critical roles in I/R induced brain injury [24]. We then performed ELISA assay to determine whether bilobilade treatment can regulate NO and VEGF levels following I/R injury to the brain. NO production was inhibited and VEGF levels were increased by bilobalide treatments (Fig. 5A,B). Since the PI3K/Akt and eNOS pathways are well established mechanisms for angiogenesis and endothelial development, additional experiments were performed in order to examine these as possible molecular targets for bilobilade (10 mg/kg) action. Western blot analysis of cerebral homogenates showed an upregulation of phosphorylated Akt and eNOS in bilobalide(10 mg/kg) treated samples (Fig. 5C,D).

Bilobalide treatment induces NO and VEGF production through activation of eNOS and Akt pathways. (A) Nitric oxide (NO) and (B) VEGF production from infarcted areas of the brain were measured by ELISA analysis. Bilobalide treatment inhibited NO and promoted VEGF production. (C) Western blot analysis showed that bilobalide (10mg/kg) treatment enhanced p-Akt and p-eNOS levels. Results were obtained from three independent experiments; only one representative experiment is shown. (D) Quantitative densitometry of western blots presented in (C). Data are presented as mean±SD. n=12 rats per group at 24 hour time point; n=10 rats per group at 7 day time point. **: p< 0.01; *: p< 0.05.
Bilobalide treatment induces NO and VEGF production through activation of eNOS and Akt pathways. (A) Nitric oxide (NO) and (B) VEGF production from infarcted areas of the brain were measured by ELISA analysis. Bilobalide treatment inhibited NO and promoted VEGF production. (C) Western blot analysis showed that bilobalide (10mg/kg) treatment enhanced p-Akt and p-eNOS levels. Results were obtained from three independent experiments; only one representative experiment is shown. (D) Quantitative densitometry of western blots presented in (C). Data are presented as mean±SD. n=12 rats per group at 24 hour time point; n=10 rats per group at 7 day time point. **: p< 0.01; *: p< 0.05.
To further explore the phenomenon that bilobalide functions by way of the Akt and eNOS pathways, specific inhibitors, LY294002 (LY) and L-NAME, respectively, were utilized. Western blot analysis of phosphorylated Akt and total eNOS was performed to confirm that treatments of LY and L-NAME can sufficiently block Akt and eNOS pathways. Brain infarct size after MCAO was increased in bilobalide (10 mg/kg) treated rats that were co-treated with either LY or L-NAME when compared to treated rats without specific inhibitors. When rats were simultaneously treated with both inhibitors, infarction size increased to almost control (non-treated) levels (Fig. 6A). Comparable changes to neurological scores assessed 24 hours and 7 days after MCAO were observed. Rats that were co-treated with either LY or L-NAME displayed greater neurological deficit than those that had been treated with bilobalide (10 mg/kg) alone, while treatment with both inhibitors resulted in deficits that were similar to control rats (Fig. 6B).These results strongly indicate that both PI3K-Akt and eNOS pathways are essential to the recovery process induced by bilobalide. Angiogenesis and cell proliferation levels, as measured by CD31 and BrdU staining, dramatically decreased in rats treated with bilobalide (10 mg/kg) and LY and/or L-NAME, with the most significant reduction found in rats treated with both inhibitors (Fig. 6C).
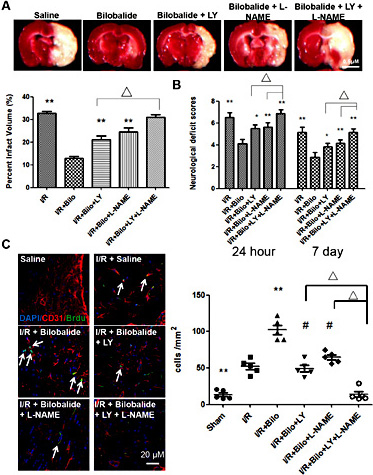
Inhibition of Akt and/or eNOS pathways diminishes the beneficial effects of bilobalide on I/R induced injury. Co-treatment with bilobalide (10mg/kg), Akt inhibitor, LY294002 (5 µl of 100 µM solution per animal), and/or eNOS inhibitor, L-NAME (5 µl of 5mM solution per animal), led to larger infarct area (A) and poorer neurological scores (B) than bilobalide treatment alone. (C) Similarly, cotreatment with LY and/or L-NAME impaired bilobalide induced angiogenesis and cell proliferation as demonstrated by CD31 and BrdU immunostaining. Data are presented as mean±SD. n=12 rats per group at 24 hour time point; n=10 rats per group at 7 day time point. ** : p< 0.01; Δ, # or * : p< 0.05. Scale bar=0.5cm.
Inhibition of Akt and/or eNOS pathways diminishes the beneficial effects of bilobalide on I/R induced injury. Co-treatment with bilobalide (10mg/kg), Akt inhibitor, LY294002 (5 µl of 100 µM solution per animal), and/or eNOS inhibitor, L-NAME (5 µl of 5mM solution per animal), led to larger infarct area (A) and poorer neurological scores (B) than bilobalide treatment alone. (C) Similarly, cotreatment with LY and/or L-NAME impaired bilobalide induced angiogenesis and cell proliferation as demonstrated by CD31 and BrdU immunostaining. Data are presented as mean±SD. n=12 rats per group at 24 hour time point; n=10 rats per group at 7 day time point. ** : p< 0.01; Δ, # or * : p< 0.05. Scale bar=0.5cm.
Similarly, the anti-apoptotic impact of bilobalide treatment was diminished by Akt and eNOS inhibition. Immunostaining revealed that the number of caspase-3 positive cells, was significantly increased in rats treated with LY and/or L-NAME in addition to bilobalide (10 mg/kg), as compared to those treated with bilobalide (10 mg/kg) alone. Consistent with previous assays, the greatest increase in caspase-3 positive cells was found in rats treated with both inhibitors (Fig. 7A). Western blot analysis showed a decreased Bcl-2/Bax ratio in the presence of LY and/or L-NAME, confirming increased levels of apoptosis. Consistent with previous data, combined treatment of LY and L-NAME showed the greatest inhibition bilobalide effects, as it resulted in the lowest Bcl-2/Bax ratio (Fig. 7B).

Co-treatment with Akt and/ or eNOS inhibitors attenuates the anti-apoptotic effects of bilobalide. Immunostaining of cleaved caspase-3 (A) and Western blot analysis of the Bcl-2/Bax ratio (B) revealed that pharmacologic inhibition of Akt and/or eNOS signaling pathways partially ablates the anti-apoptotic effects of bilobalide following I/R injury to the brain. Data are presented as mean±SD. n=12 rats per group at 24 hour time point; n=10 rats per group at 7 day time point. ## or ** : p< 0.01; Δ or # : p< 0.05.
Co-treatment with Akt and/ or eNOS inhibitors attenuates the anti-apoptotic effects of bilobalide. Immunostaining of cleaved caspase-3 (A) and Western blot analysis of the Bcl-2/Bax ratio (B) revealed that pharmacologic inhibition of Akt and/or eNOS signaling pathways partially ablates the anti-apoptotic effects of bilobalide following I/R injury to the brain. Data are presented as mean±SD. n=12 rats per group at 24 hour time point; n=10 rats per group at 7 day time point. ## or ** : p< 0.01; Δ or # : p< 0.05.
Akt and eNOS inhibitors had the same effect when measuring autophagy after ischemia. In rats treated with LY and/or L-NAME, in addition to bilobalide (10 mg/kg), there was an increase in the number of autophagic vacuoles. When treated with both LY and L-NAME, the number of autophagic vacuoles increased dramatically to levels similar to those of rats that were not treated with bilobalide (Fig. 8A). This suggests that the combined effect of the inhibitors almost completely blocked the ability of bilobalide to decrease autophagy. This was further supported by examining Beclin-1 levels and the LC3-II/LC3-I ratio. As with earlier results, both Beclin-1 levels and the LC3-II/LC3-I ratio increased in presence of either inhibitor, but the the most profound increase occurred with treatment of both LY and L-NAME (Fig. 8B). Confocal microscopy analysis of LC3-II immunostaining also showed increased autophagy levels, as punctation of LC3-II vesicles was dramatically increased in the presence of LY and/or L-NAME following ischemia. Interestingly, in contrast to previous data, the amount of punctation in rats treated with both inhibitors was not significantly greater than those treated with one or the other (Fig. 7C).
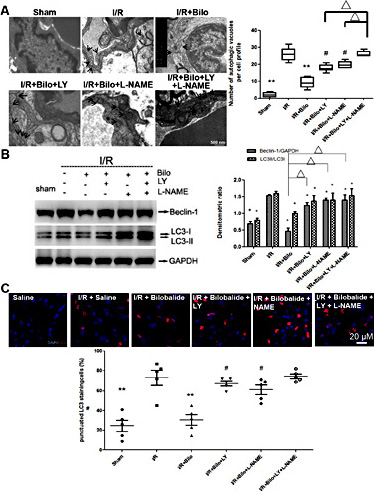
Co-treatments with Akt and/or eNOS inhibitors attenuates the anti-autophagic effects of bilobalide. Electronic microscope images of autophagic vacuoles (A), immunoblotting of Beclin-1 and LC3-II/LC3-1 ratio (B) and immunostaining of LC3 foci (shown in red) (C) revealed that pharmacologic inhibition of Akt and/or eNOS signaling pathways partially ablates the anti-autophagic effects of bilobalide (10mg/kg) following I/R injury to the brain. Data are presented as mean±SD. n=12 rats per group at 24 hour time point; n=10 rats per group at 7 day time point. ** : p< 0.01; *, Δ or # : p< 0.05.
Co-treatments with Akt and/or eNOS inhibitors attenuates the anti-autophagic effects of bilobalide. Electronic microscope images of autophagic vacuoles (A), immunoblotting of Beclin-1 and LC3-II/LC3-1 ratio (B) and immunostaining of LC3 foci (shown in red) (C) revealed that pharmacologic inhibition of Akt and/or eNOS signaling pathways partially ablates the anti-autophagic effects of bilobalide (10mg/kg) following I/R injury to the brain. Data are presented as mean±SD. n=12 rats per group at 24 hour time point; n=10 rats per group at 7 day time point. ** : p< 0.01; *, Δ or # : p< 0.05.
Discussion
Since apoptosis and autophagy are now relatively well defined mechanisms of stroke mediated brain injury [25, 26], readily available compounds that are able to combat these pathways are desirable subjects of further studies to develop therapeutic strategies to treat stroke patients. In this study, we subjected rats to transient MCAO and studied the effects of bilobalide in post MCAO recovery. Multiple benefits were observed in bilobalide treated rats including reduction of infarct size as well as improvement neurological function. These benefits were attributed to increased angiogenesis and suppressed apoptosis and autophagy by bilobalide. Reduction of apoptosis and autophagy was observed following I/R injury in rats subject to bilobalide treatment evidenced by reduced levels of caspace-3, beclin-1 and LC3-II/LC3-I ratio. These protective effects were examined and determined by inhibition of cell death mechanisms via manupulation of the Akt and eNOS survival pathways. Treatment with specific inhibitors LY294002 and L-NAME in combination with bilobalide resulted in a markedly decreased amount of cell proliferation and protection after MCAO, suggesting that bilobalide does in fact regulate apoptosis and autophagy in an Akt and eNOs dependent manner. Furthermore, treatment with both of these inhibitors simultaneously showed a stronger inhibition on bilobalide than with one inhibitor alone, suggesting that bilobalide is able to stimulate both survival mechanisms. These results were also reflected in the lower neurological scores in rats treated with both inhibitors at once.
Even though this study provides substantial evidence for upregulated Akt and eNOS being the major pathways leading to bilobalide induced cell protection and recovery, its effect on other pathways remain to be further explored. The activation of other pathways can trigger cell survival mechanisms as well. The collective group of prosurvival kinase pathways is termed reperfusion injury salvage kinase, or the RISK pathway [27]. Since bilobalide is able to modulate Akt, a component in the RISK pathway, further exploration of other prosurvival kinases such as Erk1/2 may prove fruitful in understanding how bilobalide functions. RISK pathway kinases have been observed to play multiple roles in both ischemic preconditioning (IPC) and postconditioning (IPOST) [27]. In short, brief and repeated episodes of I/R is able to “condition” the body for prolonged episodes of ischemia and help in the recovery process before and after infarction. As such, induction of PI3-Kinase, a well-established activator of Akt has been found to be critical in both forms of conditioning [28-30]. Short bouts of I/R resulted in activation of Akt in rat hearts and reduction of infarct size [29]. When combined with the fact that pharmacological activation of the RISK pathway has been found to protect against myocardial infarction [28, 31], bilobalide treatment may serve as a substitute for IPC and IPOST, providing the same resistance to infarction without suffering short episodes of I/R. This suggests another mechanism of bilobalide mediated protection against stroke; not only has it been shown to facilitate stroke recovery, but it may also serve as a pre-emptive deterrent to the most common pathways of cell death as a result of stroke. The advantage of bilobalide in this regard is the fact that it is an easily obtainable compound from Ginko Biloba and is already used in traditional eastern diet. If this therapeutic strategy will be further developed, it could be easily implemented as an alternative for IPC and IPOST that can provide similar protective qualities without having to undergo short episodes of ischemia.
Our finding that bilobalide is able to reduce apoptosis, autophagy and promote cell proliferation may be directly relevant in disrupting traditional cell death pathways as a result of ischemic injury. Treatment of bilobalide exhibits improvement in neurological scores, reduction of infarct size and is further supported by a reversal in cell apoptosis and autophagy. Our study elucidated, at least in part, the molecular action of Bilobalide and how it can protect against ischemia mediated cell death. While the model of infarction used in this study was performed in the brain, the pathways examined in this study can be examined in other organs, for example, similar mechanisms of apoptosis and autophagy are observed in myocardial infarctions [27]. The ability of bilobalide to induce cell survival via the activation of Akt and eNOS pathways can undoubtedly be applied to a wide variety of applications. As such, this study can be further broaden to study the potential beneficial effect of bilobalide on other organ injuries induced by ischemia reperfusion injuries, such as heart, kidney and lung.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No 81073087), Joint innovation project of China Academy of Chinese Medical Sciences (No ZZ11-026), National Program on Key Basic Research Project (973 Program, No 2015CB554405), National Institutes of Health (R01AR067766, R01HL124122) and American Heart Association (12SDG12070174).
YZ designed and performed experiments, analyzed data and wrote the paper. ZW and FY performed experiments and wrote the paper. MY, BY and XF performed experiments. YZ, JL and HZ developed the concept, designed experiments, analyzed data and edited the article. All authors have reviewed the manuscript and approved for submission.
Disclosure Statement
The authors declare to have no competing financial interests.
References
Y. Zheng, Z. Wu and F. Yi contributed equally to this work

