

Abstract
Background: Streptozotocin (STZ) has served as an agent to generate an Alzheimer's disease (AD) model in rats, while edaravone (EDA), a novel free radical scavenger, has recently emerged as an effective treatment for use in vivo and vitro AD models. However, to date, these beneficial effects of EDA have only been clearly demonstrated within STZ-induced animal models of AD and in cell models of AD. A better understanding of the mechanisms of EDA may provide the opportunity for their clinical application in the treatment of AD. Therefore, the purpose of this study was to investigate the underlying mechanisms of STZ and EDA as assessed upon electrophysiological alterations in CA1 pyramidal neurons of rat hippocampal slices. Methods: Through measures of evoked excitatory postsynaptic currents (eEPSCs), AMPAR-mediated eEPSCs (eEPSCsAMPA), evoked inhibitory postsynaptic currents (eIPSCs), evoked excitatory postsynaptic current paired pulse ratio (eEPSC PPR) and evoked inhibitory postsynaptic current paired pulse ratio (eIPSC PPR), it was possible to investigate mechanisms as related to the neurotoxicity of STZ and reductions in these effects by EDA. Results: Our results showed that STZ (1000 µM) significantly inhibited peak amplitudes of eEPSCs, eEPSCsAMPA and eIPSCs, while EDA (1000 µM) attenuated these STZ-induced changes at holding potentials ranging from -60mV to +40 mV for EPSCs and -60mV to +20 mV for IPSCs. Our work also indicated that mean eEPSC PPR were substantially altered by STZ, effects which were partially restored by EDA. In contrast, no significant effects upon eIPSC PPR were obtained in response to STZ and EDA. Conclusion: Our data suggest that STZ inhibits glutamatergic transmission involving pre-synaptic mechanisms and AMPAR, and that STZ inhibits GABAergic transmission by post-synaptic mechanisms within CA1 pyramidal neurons. These effects are attenuated by EDA.
Introduction
Alzheimer's disease (AD) is a neurodegenerative condition and the most common cause of dementia [1]. According to the World Alzheimer's Report, 46.8 million people were affected by AD worldwide in 2015, a number which is expected to increase to 131.5 million in 2050. The financial costs associated with AD are immense and will continue to rise with increases in the aging population [2,3].
Intracerebroventricular (ICV) administration of streptozotocin (STZ) in rats induces deregulation of brain insulin signaling and abnormalities in cerebral glucose utilization/metabolism [4,5,6], which are then accompanied by neuropathological and biochemical changes similar to those observed in sporadic AD, including cognitive dysfunction, decreased choline acetyltransferase activity, increased oxidative stress and hyperphosphorylation of tau protein [4,7,8,9,10,11,12,13,14,15,16,17]. Similar changes are also found in STZ-induced diabetic model through intraperitoneal injection or tail vein injection [18,19]. As oxidative imbalance represents a manifestation of AD, even preceding Aβ deposition and neurofibrillary tangles [20,21,22,23], ICV-STZ treated rats could serve as a model of AD. We have demonstrated previously that STZ was effective in inducing neurotoxicity upon synaptic transmission [24], but much work remains to be done with regard to these STZ-induced effects in CA1 pyramidal neurons of rat hippocampal slices.
Recent findings have indicated that edaravone (3-methyl-1-phenyl-2-pyrazolin-5-one, EDA), a free radical scavenger, represents a particularly intriguing drug due to its unique characteristics and success in the treatment of patients and in animal models of various diseases. Not only has EDA been shown to be effective and safe in the clinical treatment of ischemia and inflammation within the brain, heart and vessels [25,26], but also shows protective effects against amyotrophic lateral sclerosis (ALS) and Parkinson's disease (PD) as reported in experimental studies [27,28,29,30,31,32,33]. Based upon these findings it has been proposed that EDA may be a potent agent in the treatment of neurodegenerative disorders associated with oxidative stress. As oxidative imbalance plays an important role in the development and neuronal deterioration associated with AD, an increasing number of studies have been directed toward assessing the therapeutic effects of EDA within in vivo or in vitro models of AD [11,34,35,36,37,38].
Emerging data continues to accrue which indicate that prior to neuronal atrophy, the early symptoms of AD correlate with deficits in neurotransmitters (e.g. glutamate and GABA) and synaptic function seems to be affected in major brain areas involved in learning and memory, which result in cognitive impairments [39,40,41,42,43,44,45,46,47]. Our study focused on hippocampal CA1 pyramidal neurons, a brain area which is critically responsible for certain types of learning and memory and is most vulnerable in AD patients [19,48].
Within our laboratory, we have demonstrated neuroprotective effects of EDA upon STZ-induced models of AD [11] and STZ-induced electrophysiological changes in CA1 pyramidal neurons of rat hippocampal slices [24]. Specifically, as revealed from measures of spontaneous excitatory postsynaptic current (sEPSC), miniature excitatory postsynaptic current (mEPSC), spontaneous inhibitory postsynaptic current (sIPSC) and miniature inhibitory postsynaptic current (mIPSC), we found that EDA could attenuate acute and chronic neurotoxicity of STZ within CA1 pyramidal neurons of rat hippocampal slices [24]. In this study, we directed our investigation toward examining the effects of EDA on the CA3-CA1 synaptic pathway in this STZ model as achieved by measuring evoked excitatory postsynaptic currents (eEPSCs), AMPAR-mediated eEPSCs (eEPSCsAMPA), evoked inhibitory postsynaptic currents (eIPSCs), evoked excitatory postsynaptic current paired pulse ratio (eEPSC PPR) and evoked inhibitory postsynaptic current paired pulse ratio (eIPSC PPR).
Materials and Methods
Solutions and drugs
Sucrose artificial cerebrospinal fluid (sucrose-ACSF) containing (in mM): 213 Sucrose, 2.5 KCl, 2 CaCl2, 2 MgSO4, 10 D-glucose, 26 NaHCO3, and 1.25 NaH2PO4 (pH 7.4-7.5) and normal ACSF containing (in mM):126 NaCl, 2.5 KCl, 2 CaCl2, 2 MgSO4, 10 D-glucose, 26 NaHCO3,and 1.25 NaH2PO4 (pH 7.4-7.5) were used in these experiments. Streptozotocin (STZ, from Sigma, St Louis, MO, USA) was dissolved in the oxygenated ACSF and prepared immediately prior to use in the experiments. Edaravone (EDA, 3-methyl-1-phenyl-2-pyrazolin-5- one, from Calbiochem, Germany) was dissolved in 1 N NaOH, diluted with ACSF and pH adjusted to 7.4 with 1 N HCl according to the manufacturer's instructions. The pipette solution for recording EPSCs contained (in mM): 140 K-Gluconate, 10 HEPES, 2 Na2ATP, 3 KCl, 2 MgCl2 and 10 EGTA, with pH adjusted to 7.3-7.4 using KOH. A pipette solution for recording IPSCs contained (in mM): 125 Cs-methanesulfonate, 10 CsCl, 2 MgCl2, 5 NaCl, 10 HEPES, 1 EGTA, 5 Mg-ATP and 0.3 Na3GTP, with pH adjusted to 7.3-7.4 using CsOH. Bicuculline (BMI, 10 µM, from Sigma, St Louis, MO, USA) was used to block gamma-aminobutyric acid type A (GABAA) receptor mediated synaptic currents. 2-amino-5-phosphonovaleric acid (AP V, 50 µM, from Sigma, St Louis, MO, USA) was used to block N-methyl-D-aspartic acid (NMDA) glutamate receptor mediated synaptic currents. 6-cyano-7-nitroquinoxaline-2, 3-dione (CNQX, 10 µM, from Sigma, St Louis, MO, USA) was used to block non-NMDA glutamate receptor mediated synaptic currents. BMI, AP V and CNQX were prepared as stock solutions in 100% dimethylsulfoxide (DMSO) and diluted directly in pre-gassed (95% O2-5% CO2) ACSF to generate the concentrations described. The final DMSO concentration was <0.1 %.
Slice Preparation
All experiments were performed in accordance to the guidelines of the Medical Experimental Animal Administrative Committee of China and all efforts were made to minimize the number of animals used and their suffering. Hippocampal slices from Wistar rats of both genders (14-18 days) were prepared as described previously [49,50]. The animals were decapitated and brains were placed in cold (0-4°C, saturated with 95% O2-5% CO2) sucrose-ACSF for 1 min. The brains were then cut into 300 µm thick slices with a vibratome (NVSLM1, WPI, USA) and slices were incubated with ACSF (saturated with 95% O2-5% CO2) at 35°C for at least 1 h.
Electrophysiological Recordings
A conventional whole cell patch clamp technique was used in the present study. One slice with the hippocampus was transferred to the recording chamber (1 ml), submerged within the ACSF and continuously superfused with ACSF at a flow rate of 2 ml/min. Slices were viewed under an infrared differential interference microscope (40×water immersion lens, Nikon, Japan). Hippocampal neurons were visualized on a television monitor connected to a low light sensitive CCD camera (1300-QD, VDS, Germany). Activity of hippocampal CA1 neurons were recorded using an amplifier (Axon 700B, Foster City, CA) and an analogue/digital interface (CED Power 1401, Cambridge, UK) in voltage-clamp mode with 1 kHz filtration and 10 kHz digitization. The series resistance was compensated 70-80% and data were discarded if the series resistance changed by more than 20%. Patch electrodes with a resistance of 4-8 MΩ were pulled using a micropipette puller (P-97, Sutter Instrument, USA) and filled with the pipette solution.
In order to collect evoked currents, a stimulating electrode (WPI, USA) was placed on the Schaffer collateral passway at 100-200 µm distal from the recorded pyramidal neuron. The stimuli consisted of single 0.1 ms, 5-10 V pulses delivered at 15 s intervals using a stimulator (L-GLQ-1, Nanjing, China). Paired pulse ratio (PPR) stimulation using the parameters described above and separated by 100 ms was used in this study to distinguish between pre- and post-synaptic mechanisms. The synaptic response was recorded at membrane potentials between -60 mV and +40 mV for EPSCs and between -60 mV and +20 mV for IPSCs in 20 mV increments. Evoked EPSCs (eEPSC) were recorded in Mg2+-free ACSF (without readjusting the concentration of the other ions) in the presence of BMI while evoked AMPA receptor-mediated EPSCs (eEPSCAMPA) were recorded in the presence of BMI and AP Ⅴ, with both EPSCs being were recorded at a holding potential of -60 mV. Evoked GABAA receptor-mediated IPSCs were recorded in the presence of AP V and CNQX at a holding potential of +20 mV.
Postsynaptic current recordings began at 5 min after membrane rupture when the current achieved a steady state. Subsequently, STZ (1, 10, 100, 1000µM) or STZ (1000µM) +EDA (1, 10, 100, 1000µM) were administered to the slices for a minimum of 5 min to detect any potential effects upon the properties of all physiological indices in the voltage-clamp mode. For the STZ +EDA group, a pre-treatment of STZ was administered for a period of 30 min prior to the application of EDA to avoid the potential for a direct interaction between STZ and EDA. Only one slice was utilized and only a single neuron was studied within each slice for any given experiment. All electrophysiological experiments were performed at room temperature (22-25°C).
Data analysis
Data were stored on a personal computer using the Signal 3.06 software and analyzed using the Graphpad prism 6.01, Origin 8.0 and SPSS 19.0. Statistical significance was determined using paired Student's t-tests (2 conditions) or one way analysis of variance (one way ANOVA, >2 conditions). All the data were presented as mean ± SEM. P < 0.05 was required for results to be considered statistically significant.
Results
As a result of our previous work indicating that the wash-out of STZ state showed no effects compared to STZ state, currents were tested in the pre-treatment of STZ for at least 30 minutes before application of EDA, which could then avoid a direct interaction between STZ and EDA and also represent a co-application of STZ and EDA [24]. Moreover, EDA alone did not affect synaptic transmission compared with the control state [24]. Accordingly, EDA-induced changes could not compensate for the STZ-induced changes.
Effects of STZ and EDA on resting membrane potential and capacitance
We evaluated characteristics of resting membrane potential and capacitance from different concentrations of STZ or STZ (1000 µM) with different concentrations of EDA, by recording from CA1 pyramidal neurons in voltage-clamp mode. As summarized in Table 1, application of STZ (1-100 µM) had no significant effects on resting membrane potential and capacitance, but STZ (1000µM) affected resting membrane potential. With pretreatment of STZ (1000µM), there were no differences in the measured electrophysiological characteristics among different concentrations of EDA (Table 2).


Effects of STZ and EDA on eEPSCs
Previously, we demonstrated that STZ (1000 µM) inhibited the amplitude and frequency of sEPSCs and mEPSCs, while EDA (1000 µM) attenuated these STZ-induced electrophysiological changes in CA1 pyramidal neurons of rat hippocampal slices. To examine further the effects of STZ and EDA on glutamatergic synaptic transmission in hippocampal slices, eEPSCs were recorded before and during STZ or EDA application.
As shown in Figure 1A, STZ (1000 µM) significantly decreased the amplitude of eEPSCs. The mean amplitude was 295.75±16.50 pA at pre-STZ determinations (n = 5). After application of STZ, it was reduced to (% of control) 98.2±2.4 at 1 µM (292.61±23.35 pA, n = 5, P =0.626), 99.0±2.0 at 10 µM (294.71.93±18.17 pA, n = 5, P =0.786), 99.0±2.0 at 10µM (294.71.93±18.17 pA, n = 4, P =0.786), 91.0±1.8 at 100 µM (276.85±17.40 pA, n = 6, P <0.05), and 70.1±4.6 at 1000 µM (242.93±11.45 pA, n = 5, P <0.01). Figure 1B (left column) shows the dose-response curve for STZ. These eEPSCs amplitudes were significantly reduced by STZ (1000 µM) at all holding membrane potentials between -60 mV and +40 mV (Fig. 1C).
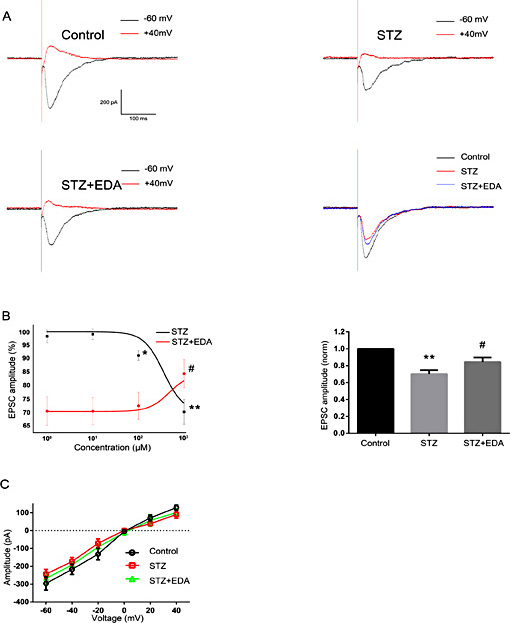
Effects of STZ and EDA on eEPSCs upon CA1 pyramidal neurons in hippocampal slices. A. Representative traces of eEPSC under control conditions and in response to STZ (1000 µM) and EDA (1000 µM). The recordings were performed at a holding potential of -60 mV. B. STZ induced a significant reduction in peak amplitudes of eEPSCs, while a partial recovery in these amplitudes was observed with EDA (*P < 0.05, **P < 0.01 versus control group, #P < 0.05, ##P < 0.01 versus STZ group). C. Effects of STZ and EDA on average current-holding potentials of one representative neuron (holding potentials ranged from -60 mV to +40 mV). Significant differences were present among these groups.
Effects of STZ and EDA on eEPSCs upon CA1 pyramidal neurons in hippocampal slices. A. Representative traces of eEPSC under control conditions and in response to STZ (1000 µM) and EDA (1000 µM). The recordings were performed at a holding potential of -60 mV. B. STZ induced a significant reduction in peak amplitudes of eEPSCs, while a partial recovery in these amplitudes was observed with EDA (*P < 0.05, **P < 0.01 versus control group, #P < 0.05, ##P < 0.01 versus STZ group). C. Effects of STZ and EDA on average current-holding potentials of one representative neuron (holding potentials ranged from -60 mV to +40 mV). Significant differences were present among these groups.
When eEPSCs were examined in the presence of EDA with pre-treatment of STZ (1000 µM), we found that reductions in amplitudes resulting from STZ were restored to (% of control) 70.4±5.4 at 1 µM (239.63±11.06 pA, n = 4, P =0.968), 70.3±5.0 at 10 µM (237.40±9.20 pA, n = 6, P =0.970), 72.3±5.0 at 100 µM (240.91±9.96 pA, n = 5, P =0.751), and 84.3±5.3 at 1000 µM (267.39±13.04 pA, n = 5, P <0.05). Figure 1B (left column) shows the dose-response curve of EDA with pre-treatment of STZ (1000 µM). Again, these results were observed at all holding membrane potentials from -60 mV to +40 mV (Fig. 1C).
Taken together, as shown in Figure 1B (right column), eEPSCs amplitudes were significantly reduced by STZ (1000 µM) and were restored by STZ (1000 µM) + EDA (1000 µM).
Effects of STZ and EDA on eEPSCsAMPA
As fast excitatory synaptic transmission is mainly mediated by AMPAR [51], in this experiment we examined the effects of STZ and EDA on postsynaptic AMPA receptors as assessed by adding AP V to the extracellular fluid.
Clear reductions in trace amplitudes were obtained from AMPAR-mediated eEPSCs (eEPSCsAMPA) between the pre-STZ and STZ conditions (Fig. 2A). The mean amplitude was 216.49±7.84 pA at pre-STZ determinations (n = 5). After application of STZ, statistical analysis revealed that the mean amplitudes of eEPSCsAMPA were decreased to (% of control) 96.4±1.3 at 1 µM (208.25±2.3 pA, n = 5, P =0.346), 96.5±1.5 at 10 µM (207.66±7.6 pA, n = 4, P =0.360), 91.5±2.0 at 100 µM (198.58±12.12 pA, n = 5, P <0.05), and 74.9±5.3 at 1000 µM (144.01±12.71 pA, n = 5, P <0.01). Figure 2B (left column) shows the dose-response curve of STZ. A representative example of an eEPSCsAMPA recorded by STZ (1000 µM) under holding membrane potentials of between -60 mV and +40 mV with its corresponding current-voltage curves is shown in Figure 2C.

Effects of STZ and EDA on eEPSCsAMPA upon CA1 pyramidal neurons in hippocampal slices. A. Representative traces of eEPSCsAMPA under control conditions and in response to STZ (1000 µM) and EDA (1000 µM). The recordings were performed at a holding potential of -60 mV. B. STZ induced a significant reduction in peak amplitudes of eEPSCsAMPA, while a partial recovery in these amplitudes was observed with EDA(*P < 0.05, **P < 0.01 versus control group, #P < 0.05, ##P < 0.01 versus STZ group). C. Effects of STZ and EDA on average current-holding potential upon one representative neuron (holding potentials ranged from -60 mV to +40 mV). Significant differences were present among these groups.
Effects of STZ and EDA on eEPSCsAMPA upon CA1 pyramidal neurons in hippocampal slices. A. Representative traces of eEPSCsAMPA under control conditions and in response to STZ (1000 µM) and EDA (1000 µM). The recordings were performed at a holding potential of -60 mV. B. STZ induced a significant reduction in peak amplitudes of eEPSCsAMPA, while a partial recovery in these amplitudes was observed with EDA(*P < 0.05, **P < 0.01 versus control group, #P < 0.05, ##P < 0.01 versus STZ group). C. Effects of STZ and EDA on average current-holding potential upon one representative neuron (holding potentials ranged from -60 mV to +40 mV). Significant differences were present among these groups.
When eEPSCsAMPA were examined in the presence of EDA, we found that amplitudes resulting from STZ were significantly restored to (% of control) 74.9±5.3 at 1 µM (239.63±11.06 pA, n = 6, P =0.987), 75.0±3.9 at 10 µM (239.63±11.06 pA, n = 5, P =0.938), 75.4±5.4 at 100 µM (239.63±11.06 pA, n = 4, P =0.985), and 89.6±4.7 at 1000 µM (239.63±11.06 pA, n = 5, P <0.05). Figure 2B (left column) shows the dose-response curve of EDA with pre-treatment of STZ (1000 µM). Similar effects of EDA (1000 µM) on STZ-induced (1000 µM) changes were observed upon the six holding membrane potentials as tested between -60 mV and +40 mV in 20 mV increments (Fig. 2C).
Taken together, as shown in Figure 2B (right column), eEPSCsAMPA amplitudes were significantly reduced by STZ (1000 µM) and were restored by STZ (1000 µM) + EDA (1000 µM).
Effects of STZ and EDA on eEPSC paired pulse ratio (PPR)
To examine further the effects of STZ and EDA in the CA3-CA1 passway, eEPSCs were recorded in response to pairs of stimuli separated by an interval of 100 ms. The eEPSC PPR was then calculated as a means to distinguish between pre- and post-synaptic mechanisms, with a change of PPR suggesting an involvement of pre-synaptic mechanisms. PPR was defined as the ratio of the amplitude of the second divided by the first EPSC. A typical example of an eEPSC PPR is contained in Figure 3A and shows that the second eEPSC was greater than the first for almost all PPRs determined within CA1 pyramidal neurons of rat hippocampal slices. Although mean peak amplitudes of STZ (1000 µM) were smaller than that of controls, mean PPR were significant increased in response to STZ (1.85±0.02) as compared with that in the pre-STZ condition (1.59±0.06; n = 5, P< 0.05, Fig. 3B). This STZ-induced increase in PPR was significantly decreased (1.69±0.05, Fig. 3A) in response to STZ (1000 µM) + EDA (1000 µM).

Effects of STZ and EDA on eEPSC PPR upon CA1 pyramidal neurons of hippocampal slices. A. Representative traces of eEPSC under control conditions and in response to STZ (1000 µM) and EDA (1000 µM) as obtained in response to paired stimuli with 100 ms intervals (holding potentials was -60 mV). B. Mean PPR among different drug applications (*P < 0.05, **P < 0.01 versus control group, #P < 0.05, ##P < 0.01 versus STZ group). Significant differences were present among these groups.
Effects of STZ and EDA on eEPSC PPR upon CA1 pyramidal neurons of hippocampal slices. A. Representative traces of eEPSC under control conditions and in response to STZ (1000 µM) and EDA (1000 µM) as obtained in response to paired stimuli with 100 ms intervals (holding potentials was -60 mV). B. Mean PPR among different drug applications (*P < 0.05, **P < 0.01 versus control group, #P < 0.05, ##P < 0.01 versus STZ group). Significant differences were present among these groups.
Effects of STZ and EDA on eIPSCs
Previous work within our laboratory also demonstrated that STZ inhibited amplitudes and frequencies of sIPSCs and mIPSCs, while EDA attenuated these STZ-induced electrophysiological changes in CA1 pyramidal neurons of rat hippocampal slices [24]. Although these findings clearly establish the neurotoxic effects of STZ and inhibitory effect by EDA, in this experiment the impact of these drugs on evoked IPSCs (eIPSCs) were assessed as a means to reveal the underlying mechanisms of these effects.
To test whether STZ modulated GABAergic synaptic transmission, we recorded eIPSCs in CA1 pyramidal neurons. During the absence or presence of STZ (1000μM), there was a significant reduction in eIPSCs amplitude in response to STZ (Fig. 4A). The mean amplitude was 283.08±9.26 pA at pre-STZ determinations (n = 5). After application of STZ, it was reduced to (% of control) 97.7±1.2 at 1 µM (277.61±10.41 pA, n = 6, P =0.626), 97.2±1.0 at 10 µM (274.90±8.48 pA, n = 6, P =0.626), 93.7±1.5 at 100 µM (276.99±6.96 pA, n = 5, P =0.626), and 73.9±3.2 at 1000 µM (225.65±7.55 pA, n = 5, P =0.626). Figure 4B (left column) shows the dose-response curve of STZ. Remarkable reductions by STZ (1000 µM) in eEPSCsAMPA amplitudes were also present at all holding membrane potentials between -60 mV and +20 mV (Fig. 4C).

Effects of STZ and EDA on eIPSC upon CA1 pyramidal neurons of hippocampal slices. A. Representative traces of eIPSC under control conditions and in response to STZ (1000 µM) and EDA (1000 µM). The recordings were performed at a holding potential of -60 mV. B.STZ induced significant reductions in peak amplitudes of eIPSCs, while a partial recovery in these amplitudes was observed with EDA(*P < 0.05, **P < 0.01 versus control group, #P < 0.05, ##P < 0.01 versus STZ group). C. Effects of STZ and EDA on average current-holding potential of one representative neuron (holding potentials ranged from -60 mV to +20 mV). Significant differences were present among these groups.
Effects of STZ and EDA on eIPSC upon CA1 pyramidal neurons of hippocampal slices. A. Representative traces of eIPSC under control conditions and in response to STZ (1000 µM) and EDA (1000 µM). The recordings were performed at a holding potential of -60 mV. B.STZ induced significant reductions in peak amplitudes of eIPSCs, while a partial recovery in these amplitudes was observed with EDA(*P < 0.05, **P < 0.01 versus control group, #P < 0.05, ##P < 0.01 versus STZ group). C. Effects of STZ and EDA on average current-holding potential of one representative neuron (holding potentials ranged from -60 mV to +20 mV). Significant differences were present among these groups.
In response to EDA with pre-treatment of STZ (1000 µM), eIPSCs amplitudes were restored to (% of control) 73.5±3.9 at 1 µM (231.55±4.66 pA, n = 5, P =0.949), 73.9±4.0 at 10 µM (224.62±9.30 pA, n = 5, P =0.992), 74.5±3.8 at 100 µM (228.28±7.92 pA, n = 4, P =0.897), and 88.5±2.9 at 1000 µM (256.93±17.68 pA, n = 5, P <0.01). Figure 4B (left column) shows the dose-response curve of EDA with pre-treatment of STZ (1000 µM). Similar effects were observed at all holding membrane potentials from -60 mV to +20 mV (Fig. 4C).
Taken together, as shown in Figure 4B (right column), eEPSCs amplitudes were significantly reduced by STZ (1000 µM) and were restored by STZ (1000 µM) + EDA (1000 µM).
Effects of STZ and EDA on eIPSC PPR
Figure 5A contains raw traces of eIPSC PPR among the three conditions as obtained from one typical neuron within the hippocampus slice. The data show that the second eIPSC was smaller than the first for almost all PPRs and mean peak amplitudes of STZ (1000 µM) were smaller than that of controls. The PPR for eIPSCs as determined at 100 ms inter-stimulus interval was not significantly altered by STZ (1000 µM) as compared with pre-STZ levels (0.76±0.06 versus 0.80±0.01; n = 6, P>0.05, Student's t test, Fig. 5B). Nor were any significant differences obtained in response to STZ (1000 µM)+EDA (1000 µM) as compared with that of the STZ condition (0.77±0.02, n = 6, P>0.05, Fig. 5B).

Effects of STZ and EDA on eIPSC PPR upon CA1 pyramidal neurons of hippocampal slices. A. Representative traces of eIPSC under control conditions and in response to STZ (1000 µM) and EDA (1000 µM) as obtained in response to paired stimuli with 100 ms intervals (holding potentials was +20 mV). B. Mean PPR among different drug applications (P>0.05).
Effects of STZ and EDA on eIPSC PPR upon CA1 pyramidal neurons of hippocampal slices. A. Representative traces of eIPSC under control conditions and in response to STZ (1000 µM) and EDA (1000 µM) as obtained in response to paired stimuli with 100 ms intervals (holding potentials was +20 mV). B. Mean PPR among different drug applications (P>0.05).
Discussion
Results from a number of studies have revealed the therapeutic effects of EDA in the treatment of AD-related cognitive impairments and neuropathologies, as revealed in animal models of AD [34,35,36,37,52,53]. Work within our laboratory has shown that the learning and memory impairments and neuropathological changes (oxidative stress, neuronal histomorphology and tau protein phosphorylation) present in the ICV-STZ induced rat model of AD were significantly improved following EDA treatment [11].While these findings demonstrate the validity of this model to induce neurotoxic effects related to AD and its inhibition by EDA, the underlying electrophysiological alterations resulting from STZ and EDA within hippocampal slices remain unknown. Therefore, in this experiment, we applied a whole-cell patch clamp technique as an approach to address this issue.
Selection of the STZ doses used in this study was based upon those used in other studies as well as preliminary work within our own laboratory [24,54,55,56,57,58]. An additional consideration waste short half-life of STZ (about 5 min) and limited period of viability of acute hippocampal slices (12-16 h). Therefore, high concentrations of STZ (1000 µM) for short exposure periods were applied in some experiments (PPR). Results from numerous studies have shown that high concentrations of EDA were required to alleviate toxicity of other agents and that these concentrations ranged from several to dozens of millimoles, which did not apparently influence results as compared the control group [37,38,59,60,61]. Results from our previous work also showed that EDA did not affect postsynaptic currents [24]. As based upon preliminary experimental results and information contained in the literature, the selection of EDA concentration was established.
With this technique it was possible to derive some novel perspectives regarding the interactions among STZ, STZ+EDA and neuronal networks. We had shown previously that STZ inhibits glutamatergic and GABAergic synaptic transmission in hippocampal CA1 neurons of rats while EDA prevents this inhibition, suggesting that, in part, these drugs were involved with functional changes involving neurotransmitter release [24]. In order to investigate further the underlying mechanisms of these alterations, we examined eEPSCs and eIPSCs in CA1 pyramidal neurons of hippocampal slices in response to STZ and EDA. The activity of eEPSCs and eIPSCs represent two experimental methods which can provide an index of information transfer via Schaffer collaterals. Although an absence of interactions between EDA and synaptic networks was verified by our previous work, the decreased amplitudes of eEPSCs and eIPSCs observed in response to STZ and attenuations of these amplitude reductions with EDA suggested that glutamatergic and GABAergic projections were adversely affected by STZ; and, that the recovery of these parameters resulting from EDA confirmed the existence of an interaction between EDA and STZ-induced injury of synaptic transmission. Moreover, the fact that fast excitatory synaptic transmission is mainly mediated by AMPAR [51], prompted us to examine the electrophysiological effects of STZ and EDA on post-synaptic AMPA receptors. The decreased eEPSCsAMPA amplitudes to STZ and its recovery with EDA indicate that postsynaptic effects of STZ and EDA might be AMPAR-dependent.
In our previous work, both the amplitudes and frequencies of mEPSCs or mIPSCs were inhibited by 1000 µM STZ [24].Miniature postsynaptic currents (mPSCs) reflect the synaptic network mediated by spontaneous fusion of vesicles. The decrease in the amplitudes of mPSCs represents a decrease in postsynaptic receptor numbers or a change of receptor reactivity, and the decrease in mPSCs frequencies can be explained by a reduction in presynaptic transmitter release probability [50,62,63]. The simultaneous decrease of both parameters cannot be described as pre- or post-synaptic mechanisms, so additional studies (e.g. PPR) are required to distinguish between these possibilities.
Interactions between STZ and EDA and excitatory or inhibitory synaptic transmission have been well established, but the exact site affected by these agents remains unknown. Hence, the effects of STZ and EDA on presynaptic neurotransmitter release were also investigated by recording PPR. PPR represents a form of short-term plasticity in the nervous system and was used as a means to distinguish between pre- and post-synaptic mechanisms, changes of which suggest the involvement of a pre-synaptic mechanism.PPR>1 indicates synapses displaying paired pulse facilitation (PPF), while PPR<1, a paired pulse depression (PPD) [64]. Most eEPSCs PPR recorded in this study had values greater than one, but the majority of eIPSCs PPR were less than one.
Amplitudes of the first and second eEPSCs were both inhibited by STZ, however, this inhibition within the second response was lower than that observed for the first, resulting in an increase in PPR. Such results suggest the possibility of a decrease in glutamate release after STZ application [65]. Moreover, the significant differences in PPR between the STZ and EDA conditions indicate an increase in glutamate release after EDA application. Therefore, the neurotoxicity of STZ and effects of EDA upon glutamatergic transmission appear to involve pre-synaptic mechanisms.
Our data also revealed that although there was a decrease in the amplitude of the first and second eIPSCs with STZ, no significant differences were obtained in PPR, indicating that there were no changes in GABA release in response to these agents and that inhibition of neurotransmission by STZ and effects of EDA upon GABAergic transmission appears to be through post-synaptic mechanisms.
Taken together, our data suggest that STZ inhibits synaptic transmission. As generally reported, STZ is a cytotoxic glucose linked to a reactive nitrosourea moriety [66]. Once this molecule enters the cell the nitrosourea moriety is released and actively poison cells by cross-linking vital structure [67]. Therefore, we propose that STZ might injure the structure of synapses acutely and directly, which might be related to cytotoxic effects of STZ (e.g. DNA methylation, nitric oxide production and free radical generation) [68]. More studies will be required to test the cytotoxic effects of STZ upon synaptic transmission.
As previous results revealed that EDA alone could not affect synaptic transmission [24], we hypothesized that the neuroprotective effects of EDA might involve its free radical scavenging ability which then attenuates effects on STZ-induced electrophysiological changes. The cytotoxicity of STZ has been shown to be associated with free radical generation and preventive effects of EDA on oxidative stress in ICV-STZ rats have been demonstrated [11]. Additional experiments need to be performed to verify the relationship between the effects of EDA observed in this study and its free radical scavenging ability.
STZ is often used to induce AD in animal models and our data demonstrate its inhibitory effects on synaptic transmission, which likely results in its cytotoxicity and leads to cognitive deficits. Impaired synaptic transmission can also be observed in initial stages of AD [40,41], indicating that this STZ-induced model provides an effective means for investigating the underlying mechanisms of development and neuronal deterioration associated with AD. Indeed, increasing evidence has accumulated in support of the concept that synaptic dysfunction and failure are involved in the numerous neurobiological and neurochemical defects that characterize the AD process. Accordingly, maintenance of synaptic transmission may represent a new and important therapeutic strategy for AD.
Conclusion
In conclusion, our results as obtained from electrophysiological recordings demonstrate that STZ induces significant neurotoxicity upon glutamatergic and GABAergic synaptic transmission, effects that were attenuated with EDA. The alterations observed upon EPSCs appear to be AMPAR-dependent and involve presynaptic mechanism, while those of IPSCs appear to involve postsynaptic mechanisms. Accordingly, the present study provides novel evidence for potential therapeutic effects of EDA in the treatment of AD, through its capacity to interact with STZ-induced injury of synaptic networks.
Acknowledgement
We are grateful to Dr. Liming Zhang for providing financial support. The funding source had no such involvement in this study. This manuscript was revised for submission in English by the ED-IT Editorial Service.
Disclosure Statement
The authors declare that there are no conflicts of interest.
References
T. Ju and Y. Li contributed equally to this work

