

Abstract
Kidney morphogenesis and patterning have been extensively studied in animal models such as the mouse and zebrafish. These seminal studies have been key to define the molecular mechanisms underlying this complex multistep process. Based on this knowledge, the last 3 years have witnessed the development of a cohort of protocols allowing efficient differentiation of human pluripotent stem cells (hPSCs) towards defined kidney progenitor populations using two-dimensional (2D) culture systems or through generating organoids. Kidney organoids are three-dimensional (3D) kidney-like tissues, which are able to partially recapitulate kidney structure and function in vitro. The current possibility to combine state-of-the art tissue engineering with clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated systems 9 (Cas9)-mediated genome engineering provides an unprecedented opportunity for studying kidney disease with hPSCs. Recently, hPSCs with genetic mutations introduced through CRISPR/Cas9-mediated genome engineering have shown to produce kidney organoids able to recapitulate phenotypes of polycystic kidney disease and glomerulopathies. This mini review provides an overview of the most recent advances in differentiation of hPSCs into kidney lineages, and the latest implementation of the CRISPR/Cas9 technology in the organoid setting, as promising platforms to study human kidney development and disease.
Introduction
Chronic kidney disease (CKD) affects 3-17% of the adult population in countries of the European Union and is characterized by the loss of functional nephrons and the development of interstitial fibrosis. In adults, CKD either develops from primary kidney diseases like glomerulonephritis, polycystic kidney disease (PKD) or appears as a secondary complication such as in diabetic or hypertensive patients. When developed before the age of 25, (early-onset), CKD is characterized by a strong genetic component, with more than 200 loci identified, uncovering 70% of the most common CKD etiologies. Treatment of CKD patients progressing towards end-stage renal disease is currently limited to dialysis and renal transplantation. Therefore, the development of efficient platforms to study and elucidate the molecular mechanisms underlying CKD is critical to provide patients with suitable and effective therapeutic options [1].
Due to their phenotypic features, human pluripotent stem cells (hPSCs), including human embryonic stem cells (hESCs) and the closely related human-induced pluripotent stem cells (hiPSCs), represent an ideal model to meet this challenge. First, hPSCs renew by themselves indefinitely in vitro, providing an inextinguishable source of cells suitable for rapid and large-scale experimental analyses. Second, they have the potential to differentiate into all adult cell linages including the kidney, providing the appropriate genomic and cellular contexts for functional studies [2].
Although, directed differentiation of hPSCs towards defined kidney cell types offers unprecedented opportunities for studying phenotypes manifested at the cellular level, this approach may be insufficient for modelling the cellular interactions occurring in the whole kidney. This barrier may be overcome by generating hPSC-derived 3D organoids in vitro. In this regard, kidney organoids represent 3D kidney-like tissue structures, which offer the possibility of reconstructing an organismal context for in vitro disease modeling and functional genetics [3].
Concurrently with the development of improved differentiation and tissue engineering protocols, 2 approaches have recently revolutionized disease modelling in hPSCs. The first consists in reprogramming patient somatic cells to a pluripotent state, which allows generating hiPSC lines capturing the whole genetic complexity of the patient of origin. The second involves introducing or correcting mutations using engineered nucleases, a technique known as gene editing. This method allows targeting any genetic modification in hPSCs for further functional analysis. Both approaches represent fundamental tools for studying the genetic bases of CKD and kidney development [4].
Here we review recent advances in renal differentiation of hPSCs and formation of kidney organoids, showing the latest progress in kidney disease modelling using tissue and genome engineering in hPSCs. The combination of these approaches is contributing to revolutionize the study of kidney disease and development.
Human Pluripotent Stem Cells
hPSCs, including hESCs and hiPSCs, have the ability to self-renew and under differentiation conditions are able to give rise to any cell type of the organism, a property known as pluripotency. hESCs are typically derived from the pluripotent inner cell mass of the blastocyst and represent a new source for the study of tissue development and disease modelling in vitro [5]. However, the ethical issue related to the need of human embryos as well as the problem of immunogenicity hamper their use in the clinics.
In 2006, by screening a large set of pluripotency-associated transcription factors in mouse fibroblasts, Takahashi and Yamanaka [6] reported that the ectopic expression of a cocktail of only 4 transcription factors (Oct4, Sox2, Klf4 and cMyc, referred to as OSKM) can convert somatic cells to a pluripotent state, generating so-called induced pluripotent stem cells. Soon after, hiPSCs were successfully generated through reprogramming human fibroblasts using the same (OSKM) [7] or a slightly different combination of factors (OCT4, SOX2, NANOG and LIN28, referred to as OSNL) [8].
Over the last decade, numerous publications have reported the generation of patient-specific hiPSCs from a variety of diseases, demonstrating the possibility to study the disease phenotype and progression in vitro by differentiating hiPSCs into the affected cell type(s) [9,10,11,12,13,14], and also to perform drug screening studies [15,16]. Indeed, several studies have proved the generation and correction of patient-specific hiPSCs from patients with genetic-based disorders, showing the recovery of the disease-associated phenotype to a normal functional phenotype [9,11,12,17,18,19,20].
With the advent of novel gene editing technologies together with recent advances on the generation of hPSC-derived tissue structures termed organoids, researchers have now a unique scenario for human genetics investigation during development and disease progression.
Generating Kidney Organoids from hPSCs
The kidney represents one of the most complex organs in terms of development, spatial organization and lineage specification. It is comprised of up to 2 million nephrons [21], arising during development by reciprocal inductive signals between the ureteric bud (UB) and the metanephric mesenchyme (MM). Specifically, during early kidney development, the primitive streak (PS) gives rise to both the anterior and posterior intermediate mesoderm (IM). Eventually, the anterior IM generates the UB giving rise to the collecting ducts, while the posterior IM generates the MM giving rise to the epithelial cell types from the glomerulus through the connecting segment, as well as the renal stromal cells [22].
Success in isolating and culturing UB and MM progenitors has been traditionally limited by their propensity to differentiate outside of their developmental niches [23]. Recently, different laboratories have succeeded in extending the lifespan of primary nephron progenitor cells (NPCs) in culture. By recapitulating the signalling the environment of NPCs, Brown et al. [24 ]developed specific culture conditions that enabled the in vitro expansion of murine NPCs up to 10 passages [24]. In another study, Tanigawa et al. [25 ]could propagate mouse or rat NPCs for 5 passages using a different protocol. More recently, Li et al. [26] successfully derived mouse and human NPC lines reporting long-term propagation for more than 110 and 50 passages, respectively, under specific 3D culture conditions. Lately, Pode-Shakked et al. [27 ]cultured human fetal kidney cells for 3 passages using a modified NPC expansion medium based on the one developed by Brown et al. [24]. Expanded human fetal kidney cells were separated into different renal cell populations by fluorescence-activated cell sorting, and analyzed for gene expression at the single-cell level, showing for the first time a preservation in culture of discrete renal progenitor cell subpopulations depicting the different stages of human renal development [27].
Alternatively, UB and/or MM progenitors could be generated de novo through directed differentiation of hPSCs using small molecules and soluble factors, mimicking the signals triggering kidney development in vivo. Using this strategy, IM, the embryonic lineage giving rise to both the UB and the MM, has been efficiently derived from hPSCs [28,29,30]. In order to generate more defined kidney progenitor populations with increased differentiation potential, a cohort of studies has successfully differentiated UB [29], MM [30,31,32,33,34,35], or both UB and MM [36,37] progenitors from hPSCs (Fig. 1a). A summary of recent published reports on the generation of hPSC-derived kidney progenitors and kidney organoids is compiled in Table 1.
Recent protocols for directed differentiation of human pluripotent stem cells into renal progenitor cells and kidney organoids
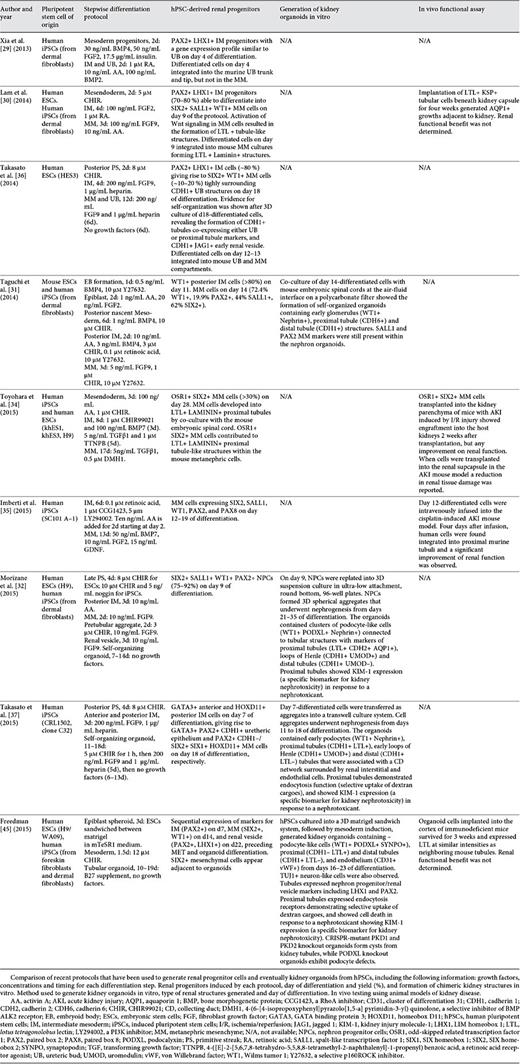
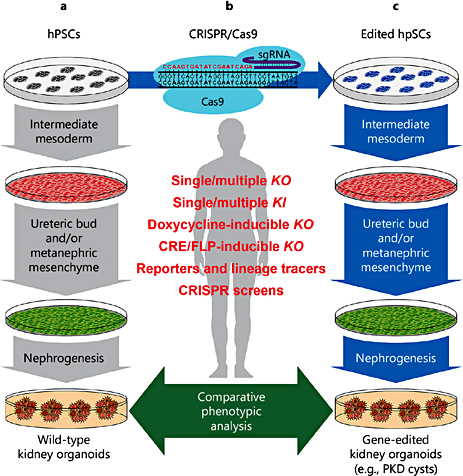
Studying kidney disease and development using genome and tissue engineering in human pluripotent stem cells. a Human pluripotent stem cells (hPSCs, grey), including hESCs and human-induced pluripotent stem cells (hiPSCs), can be differentiated towards kidney organoids using small molecules and soluble factors mimicking the signals triggering kidney development in vivo. hPSCs are first directed to intermediate mesoderm (IM, red), the embryonic lineage giving rise to both ureteric bud (UB, green) and metanephric mesenchyme (MM, green) progenitors. Depending on the protocol, UB and/or MM are then induced using different combinations of molecules. Kidney organoids (red, orange and yellow clusters) are obtained either by co-culturing UB and/or MM progenitors with mouse embryonic kidney progenitors or spinal cords. Alternatively, 100% hPSC-derived kidney organoids can now be obtained from in vitro-differentiated MM or a mixture of in vitro-differentiated MM and UB progenitors. b Genome engineering can be efficiently achieved in hPSCs using CRISPR/Cas9, which is composed by a nuclease Cas9 (blue) and a small guide (sg) RNA (red and purple) determining binding specificity trough Watson-Crick base pairing (red sequence) to its genomic target (black sequence). This approach allows introducing any modification useful for disease modelling and developmental studies in hPSCs including, single and multiple knockout (KO) or knockin (KI) mutations; creation of doxycycline-, CRE- or FLP-inducible KO lines; reporters and lineage tracer lines; and performing genetic screens using Cas9 and pooled sgRNA libraries. c The resulting engineered hPSCs (blue) can be used to study the function of developmentally or therapeutically relevant genes at each step of the differentiation towards kidney organoids and in more differentiated kidney cell types present in mature organoids.
Studying kidney disease and development using genome and tissue engineering in human pluripotent stem cells. a Human pluripotent stem cells (hPSCs, grey), including hESCs and human-induced pluripotent stem cells (hiPSCs), can be differentiated towards kidney organoids using small molecules and soluble factors mimicking the signals triggering kidney development in vivo. hPSCs are first directed to intermediate mesoderm (IM, red), the embryonic lineage giving rise to both ureteric bud (UB, green) and metanephric mesenchyme (MM, green) progenitors. Depending on the protocol, UB and/or MM are then induced using different combinations of molecules. Kidney organoids (red, orange and yellow clusters) are obtained either by co-culturing UB and/or MM progenitors with mouse embryonic kidney progenitors or spinal cords. Alternatively, 100% hPSC-derived kidney organoids can now be obtained from in vitro-differentiated MM or a mixture of in vitro-differentiated MM and UB progenitors. b Genome engineering can be efficiently achieved in hPSCs using CRISPR/Cas9, which is composed by a nuclease Cas9 (blue) and a small guide (sg) RNA (red and purple) determining binding specificity trough Watson-Crick base pairing (red sequence) to its genomic target (black sequence). This approach allows introducing any modification useful for disease modelling and developmental studies in hPSCs including, single and multiple knockout (KO) or knockin (KI) mutations; creation of doxycycline-, CRE- or FLP-inducible KO lines; reporters and lineage tracer lines; and performing genetic screens using Cas9 and pooled sgRNA libraries. c The resulting engineered hPSCs (blue) can be used to study the function of developmentally or therapeutically relevant genes at each step of the differentiation towards kidney organoids and in more differentiated kidney cell types present in mature organoids.
Xia et al. [29 ]developed a protocol where UB progenitors were obtained in 4 days by first promoting mesoderm formation in 2D hPSC cultures using bone morphogenetic protein 4 (BMP4) and fibroblast growth factor 2 (FGF2) followed by transient induction of IM cells with retinoic acid (RA), Activin A (AA) and BMP2. The resulting cells upregulated UB markers [29].
Otherwise, a number of protocols for directed differentiation of hPSCs into MM progenitors have mainly relied upon the WNT agonist CHIR99021 to induce PS cells followed by renal commitment [30,31,32,33,34,36,37]. Works from Lam et al. [30], Toyohara et al. [34] and Takasato et al. [36,37] attempted to induce SIX2 positive MM progenitors from hPSCs through the induction IM cells expressing PAX2, LHX1, and OSR1. However, the efficiency of SIX2 positive MM cells generated was relatively low (less than 33%) [30,34,36,37]. Importantly, Taguchi et al. [31], making use of lineage tracing techniques in mice, could prove that the origin of functional kidneys, the metanephros, was restricted to the posterior area of the IM where Osr1 and Wt1 were expressed, but not Pax2 and Lim1 (LHX1 in humans) [31]. These findings would anticipate that the derivation of OSR1+ WT1+ PAX2- LHX1- posterior IM cells from hPSCs might enhance the generation of SIX2 positive MM cells. Following this strategy, Taguchi et al. [31] derived MM progenitors in 14 days, showing a significant improvement on the differentiation efficiency (62% of SIX2 positive cells). Briefly, Taguchi's protocol induced 3D hPSC cultures, so-called embryoid bodies, using BMP4 treatment followed by AA, FGF2 and then exposure to a high concentration of CHIR99021, and BMP4, finally complemented with AA and RA until day 11. The last step produced MM progenitors, through CHIR99021 and FGF9 treatment until day 14 [31].
Notably, Morizane et al. [32] established a stepwise protocol capable of generating SIX2+ SALL1+ WT1+ PAX2+ NPCs, via previous induction of WT1+ HOXD11+ PAX2- LHX1- posterior IM cells, achieving 75-92% efficiency of SIX2-positive cells in only 9 days. In this case, 2D hPSC cultures were first treated with CHIR99021 followed by AA, leading to posterior IM formation at day 7. MM progenitors were then induced using FGF9 [32].
Interestingly, Takasato et al. [36,37] induced a mixture of UB and MM progenitors by adjusting the duration of CHIR99021 treatment prior to FGF9 addition, finding that a shorter CHIR99021 treatment (2 days) differentiated PS cells into anterior IM cells expressing GATA3, while a longer CHIR99021 treatment (5 days) produced posterior IM cells expressing HOXD11. Hence, CHIR99021 treatment of an intermediate duration (3-4 days) resulted in the simultaneous formation of both anterior and posterior IM cells, which at day 18 of the protocol, upon FGF9 exposure, differentiated into GATA3+ PAX2+ CDH1+ ureteric epithelium and PAX2+ CDH1- MM progenitors. These findings were in accordance with Taguchi's protocol who achieved MM induction by treating hPSCs with CHIR99021 for an extensive period of 6 days. In addition, Takasato's work showed that RA promoted the anteriorization of the IM while AGN193109 (an RA receptor antagonist) inhibited it. This was in agreement with the work of Xia et al. [29], who used RA to induce the UB fate.
In 2 recent studies, renal progenitor cells derived from hPSCs were transplanted into mice with acute kidney injury (AKI) to assess their therapeutic potential [34,35]. Specifically, Imberti et al. [35] induced hPSC-derived MM progenitors in 12 days and, although they did not test the ability of hiPSC-derived MM progenitors to further differentiate into kidney organoids in vitro, they proved for the first time that the generated renal progenitors could engraft into damaged tubules and restore renal function in a cisplatin-induced AKI mouse model. Variably, Toyohara et al. [34] transplanted hiPSC-derived OSR1+ SIX2+ renal progenitors into the kidney parenchyma of mice with ischemia/reperfusion-induced AKI, showing their engraftment into the host kidney without improvement on renal function. Interestingly, when the same cells were transplanted into the renal subcapsule, reduction in renal tissue damage was observed, suggesting a possible paracrine effect from hiPSC-derived renal progenitors [34]. Future studies are required in order to elucidate the direct or indirect therapeutic mechanisms when using hPSC-derived renal progenitors in vivo.
During kidney development, the initiation of nephrogenesis involves a mesenchymal to epithelial transition (MET) of the MM cell condensates located around the UB tip to form the renal vesicle, giving rise to the nephrons [22]. Canonical Wnt signalling directed by Wnt9b/β-catenin appears to be the dominant pathway inducing this MET process [22]. To date, only few studies have been able to recapitulate the process of nephrogenesis in vitro using hPSC-derived kidney progenitors, demonstrating their ability to self-organize into nephron-like tubular structures de novo, namely, kidney organoids (Table 1) [31,32,33,37].
Taguchi et al. [31] were the first to demonstrate the generation of kidney organoids containing nephron-like tubular structures from hPSCs. They used mouse embryonic spinal cords from embryonic day (E) 11.5 or E12.5 embryos to induce the MET of hPSC-derived MM aggregates cultured at the air-fluid interface on a polycarbonate filter. They demonstrated robust tubulogenesis showing renal structures containing nephron-like components: WT1+ Nephrin+ podocyte-like progenitor cells from the glomeruli, CDH6+ proximal tubular-like cells and CDH1+ distal tubular-like cells. However, the expression of MM markers including SALL1 and PAX2 within these renal structures was still persistent, indicating their immature state [31].
In other 3 recent studies, nephron organoids were formed in the absence of mouse embryonic spinal cords [32,33,37]. Morizane et al. [32] developed a protocol to produce kidney organoids using a 96-multiwell 3D culture system suitable for future high-throughput screening studies. Upon WNT stimulation using CHIR99021, hPSC-derived NPCs underwent MET, developing kidney organoids that contained segmented nephron-like structures expressing markers of early podocytes (WT1+ PODXL+ Nephrin+), proximal tubules (LTL+ CDH2+ AQP1+), loops of Henle (CDH1+ UMOD+) and distal tubules (CDH1+ UMOD-) [32]. Each of these regions appeared to be connected in that order, forming a nephron-like unit, as similarly described by Taguchi et al. [31]. However, in contrast to Taguchi's study, the expression of MM markers was not observed within the kidney organoids generated in this case, suggesting the formation of more mature nephron-like tubular structures [32].
Takasato et al. [37] used a transwell system to culture hPSC-derived IM cells as aggregates at the air-liquid interface. After WNT activation using CHIR99021, cell aggregates underwent nephrogenesis, revealing the formation of nephron components that included distal (CDH1+ LTL-) and proximal (CDH1+ LTL+) tubules, early loops of Henle (CDH1+ UMOD+) and glomeruli containing early podocytes (WT1+ Nephrin+) [37]. These kidney organoids also contained GATA3+ CDH1+ cell structures located at the bottom of the organoid, suggesting the formation of a presumptive collecting duct-like network [37]. However, further studies are needed to characterize in depth the identity of such structure. In addition, the existence of renal interstitial cells (FOXD1+ MEIS1+/FOXD1- MEIS1+) and endothelial cells (CD31+ KDR+ SOX17+) within these organoids was also described, indicating a considerable degree of tissue complexity [37]. Nevertheless, the gene expression profile of these kidney organoids was similar to that of first-trimester human kidneys, therefore representing immature early stage kidney organoids [37].
Following a completely distinct approach, Freedman et al. [33] used a 3D culture system consisting of culturing hPSCs between 2 layers of matrigel, referred to as matrigel sandwich, to generate epiblast spheroids expressing pluripotency-associated genes. Renal differentiation was initiated from these epiblast spheroids by high dose CHIR99021 treatment for 1.5 days to induce mesoderm specification, then followed by stochastic differentiation until day 16 [33]. Segmented nephron-like kidney organoids with podocyte-like cells (WT1+ PODXL+ SYNPO+), proximal tubules (LTL+) and endothelium (CD31+ vWF+) were generated [33]. However, TUJ1+ neuron-like cell clusters were also detected surrounding the nephron structures [33]. In addition, early markers such as LHX1 and PAX2 were still found in the tubular structures indicating the presence of immature cell phenotypes [33].
Thus, following different strategies, Taguchi's, Morizane's, Takasato's and Freedman's studies succeeded in the generation of kidney organoids from hPSCs in vitro, describing similar types of 3D renal structures [31,32,33,37]. Notably, the proximal tubule-like structures produced in Morizane's, Takasato's and Freedman's studies were able to respond to nephrotoxicity assays, indicating that the generated kidney organoids possessed a certain degree of functional maturity [32,33,37].
As a complementary assay to assess the identity and renal potential of hPSC-derived kidney progenitors, some studies have also evaluated their ability to form kidney chimeric structures in vitro by performing re-aggregation assays with mouse embryonic kidney cells (Table 1) [29,30,34,36]. In general, E11.5 mouse kidney rudiments are dissociated into single cells, mixed with hPSC-derived renal progenitors, and then cultured under organotypic conditions. After 4-6 days of culture, human cells found to be incorporated into the mouse renal structures should express specific renal epithelial markers. Using this approach, Xia et al. [29] and Takasato et al. [36] tested the capability of hPSC-derived UB and/or MM progenitors to integrate into mouse UB and/or MM renal structures. Consistently, while Xia et al. [29] only observed chimeric UB structures [29], Takasato's progenitors integrated in both UB and MM structures [36].
Despite the valuable advances, hPSC-derived kidney organoids are still not comparable to mature renal tissue. Major limitations of current kidney organoid technology are due to the lack of important kidney anatomical features, including the formation of the ureter, the generation of a proper ureteric tree connected to all collecting ducts stemming from the nephrons, and the formation of glomerular capillary loops necessary for blood filtration. Indeed, the lack of vascularization impedes organoid growth and may influence its maturation. Current approaches to promote vascularization rely on the co-culture with endothelial cells [38] and the transplantation of kidney organoids under mouse kidney capsule to induce vascularization by the host [31,39]. Of note, hPSC-derived kidney organoids may benefit from novel bioengineering approaches to re-create with fidelity the hierarchical architecture of the kidney in vitro by means of biomimetic biomaterials, micro/nanotechnology, microfluidic devices and bioreactors [40,41]. Recently, organ decellularization has opened new perspectives in tissue engineering by providing organ-specific scaffolds that may allow transplanted cells to retain their phenotypic properties, and enhance their maturation [42]. To date, the usage of acellular kidney extracellular matrix promoting differentiation of hPSCs has been reported in few studies, taking advantage of rhesus monkey kidney matrices [43,44].
In addition, present protocols for derivation of renal progenitors from hPSC do not address the necessity to expand these progenitors in vitro while preserving their nephrogenic potential [30,31,32,33,34,36,37]. In recent works by Brown et al. [24], Tanigawa et al. [25] and Li et al. [26,] hPSC-derived NPCs have only reported limited expansion under specific culture conditions that have increased the propagation of primary murine [24,25,26], rat [25] and human NPCs [26]. Thus, culture conditions for long-term expansion of hPSC-derived NPCs remain still unknown.
Overall, hPSC-derived kidney organoids provide promising models for studying both the cellular and multi-cellular phenotypes involved in kidney development and disease (Fig. 1a). To address gene function using these platforms, hPSC lines carrying mutations relevant for kidney development and disease need to be generated. This goal can currently be achieved using 2 different approaches: reprogramming and clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated systems 9 (Cas9) genome editing.
Studying the Genetic Bases of Kidney Disease and Development Using hPSCs
The discovery of hiPSCs has allowed generating cohorts of hiPSC lines carrying genetic mutations relevant to numerous kidney diseases, including PKD, renal cysts and diabetes syndrome (RCAD/MODY5), Alport syndrome, Wolfram syndrome, Wilms tumour, focal segmental glomerulosclerosis and systemic lupus erythematosus. Importantly, in some cases, the disease-relevant cell types generated by differentiation of patient-derived hiPSCs have shown to recapitulatedisease-specificfeatures in vitro [45].
In this regard, Freedman et al. [46] identified a key biological defect relevant to kidney pathophysiology in hiPSCs derived from patients with autosomal dominant PKD (ADPKD). The authors reprogrammed patient cells carrying heterozygous loss-of-function mutations in the PKD1 gene encoding polycystin-1 (PC1). Compared to wild type controls, ADPKD hiPSCs and their derivatives expressed lower levels of PC2 in the primary cilium, suggesting that PC1 may regulate PC2 protein expression in this sensory organelle, known to be crucial for PKD etiology [46].
Even though patient-derived hiPSCs have proved valuable for kidney disease modeling, the standard reprogramming protocols used often produce hiPSC lines showing significant phenotypic variation. Such variation might be a disadvantage when trying to reproduce a disease phenotype in a culture dish in vitro. To solve this problem, isogenic hPSCs differing only in the disease-causing mutation represent an attractive alternative. Such mutations can be efficiently introduced in hPSCs using Zinc-Finger nucleases, transcription activator-like effector nucleases and CRISPR/Cas.
CRISPR/Cas9 encodes 2 distinct DNA cleavage and binding modules: the endonuclease Cas9 and a single guide RNA (sgRNA). Cas9 binding specificity is determined through standard Watson-Crick base pairing of 20 nucleotides (nt) of the sgRNA to a complementary 20 nt genomic sequence (protospacer). The only restriction is that the protospacer needs to be followed in 5 prime (5′) end by an NGG motif (protospacer-associated motif or PAM, where N can be A, T, G, or C; Fig. 1b) [47]. Binding of the Cas9:sgRNA complex to a PAM-flanked protospacer induces a DNA double-strand breaks 3 bp upstream of the PAM sequence triggering DNA repair through alternative pathways: error-prone non-homologous end joining and microhomology-mediated end joining, which can lead to the creation of small insertion and deletion mutations (Indels); or homology-directed repair, leading to error-free DNA repair, or the introduction of precise nucleotide alterations when exogenous homologous DNA repair templates are provided [48]. Due to its simplicity, CRISPR/Cas9 has been widely adopted, offering a powerful and versatile tool for genome engineering in hPSCs [49].
Illustrating the power of CRISPR/Cas9 for functional human genetics, a recent study by Zhu et al. [50] combined differentiation of hPSCs and gene editing to systematically study the role of 8 pancreatic transcription factors during pancreatic development and disease. The study uncovers the specific developmental step(s) affected by mutations in these genes and highlights the importance of developing human-specific models for studying human genetics [50].
Recent advances in genome-editing technologies offer the possibility to perform sophisticated genetic studies in hPSC-derived cell types. The current challenge is to develop better approaches to re-create, as closely as possible, what occurs in a human embryo or an adult organ. A promising approach consists of studying disease-relevant mutations in organoids, which now can be efficiently derived from hPSCs. Following this rationale, Freedman et al. [33] tested the function of 3 disease-relevant genes in kidney organoids. They inactivated podocalyxin (PODXL), PKD1 and PKD2 by transiently transfecting undifferentiated hPSCs with plasmids expressing Cas9 and sgRNAs targeting these genes. Upon clonal expansion of biallelic knockout lines, the authors further differentiated the targeted hPSC lines for functional analysis. In their hands, PODXL-defective kidney organoids showed junctional organization defects in podocyte-like cells, while PKD1 or PKD2 knockouts induced cyst formation from kidney tubules. Similar defects were not observed in the epiblast-stage hPSC spheroids, suggesting that kidney organoids could recapitulate bona fide tissue-specific phenotypes [33].
Therefore, the seminal study by Freedman et al. [33] suggests that kidney organoids can recapitulate features of glomerular and tubular diseases upon experimental injury or through CRISPR/Cas9 genetic engineering. Further development and improvements of the CRISPR/Cas9 toolbox in hPSCs will certainly contribute to address fundamental questions related to kidney development and disease. For instance, the generation of inducible knockout hPSC lines will enable studying gene function in kidney organoids in a temporal and cell-specific manner. In addition, the generation of knockin reporter lines will be particularly useful for real-time observation of gene-expression, cell-lineage tracing, differentiation screens and generally for sorting pure cell population for further molecular characterization (Fig. 1c).
Conclusions and Future Directions
A major limitation of classical hPSC differentiation protocols lies in their inability to generate complex and organized 3D tissues. Recent success in the generation of hPSC-derived 3D kidney organoids has opened the door to the possibility of modeling, at least in part, the complexity and function of the human kidney in vitro. However, further improvements are necessary to generate mature functional nephron structures with vascularized glomeruli, and connected to a collecting duct system. In this regard, novel bioengineering approaches using decellularized matrices, synthetic materials and co-culture techniques could contribute to enhance the maturation of 3D kidney structures by providing controlled microenvironments [40,41,42].
On the other hand, the power and the versatility of CRISPR/Cas9 combined with the physiological properties of kidney organoids will also allow studying complex genetic diseases affecting the kidney. For instance, clear-cell renal cell carcinoma exhibits substantial mutation heterogeneity. Discriminating drivers from passenger mutations and their contribution to tumour phenotypes is still challenging, particularly in tumours that can be only modelled and studied using patient-derived xenografts [51]. In this context, systematic analysis of CRISPR/Cas9-edited patient mutations in kidney organoids represents a promising approach to functionally dissect the role of these mutations, offering ideal platforms to perform drug and genetic screens in order to identify novel therapeutic targets.
Hopefully, CRISPR-editing in hiPSCs derived from patient affected by hereditary CKD and further generation of disease-corrected organoids for autologous transplantation will soon become a realistic alternative to dialysis and renal transplantation. To become real, ethical and safety issues related to both reprogramming and CRISPR-editing need to be solved. In this regard, efforts in the generation of new Cas9 variants with reduced off-target effects together with high-stringency criteria for sgRNA design will have a strong impact for transitioning from bench to bed-side using hiPSC-derived kidney organoids.
Acknowledgements
This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (StG-2014-640525_REGMAMKID) to E.G. and N.M., F.G. is supported by the Ramon y Cajal Grant - Biomedicine (RYC-2014-16751) from the Ministry of Economy and Competitivity (MINECO), Spain. N.M. is also supported by CardioCel (TerCel, Instituto de Salud Carlos III), MINECO (SAF2014-59778, SAF2015-72617-EXP, RYC-2014-16242) and 2014 SGR 1442. We are grateful to members of the Montserrat laboratory for insightful discussions and critical reading of the manuscript.
Disclosure Statement
The authors have no conflicts of interest to declare.
