

Abstract
Increased risk for bacterial superinfections substantially contributes to the mortality caused by influenza A virus (IAV) epidemics. While the mechanistic basis for this lethal synergism is still insufficiently understood, immune modulation through the viral infection has been shown to be involved. Since the pattern-recognition receptor (PRR) toll-like receptor 7 (TLR7) is a major sensor for the viral genome, we studied how IAV recognition by TLR7 influences the development of secondary pneumococcal infection. In a mouse model of IAV, TLR7-deficient hosts induced a potent antiviral response and showed unchanged survival. In secondary pneumococcal infection during acute influenza, TLR7ko mice showed a fatal outcome similar to wild-type (WT) hosts, despite significantly delayed disease progression. Also, when bacterial superinfection occurred after virus clearance, WT and TLR7-deficient hosts showed similar mortality, even though we found the phagocytic activity of alveolar macrophages isolated from IAV-pre-infected hosts to be enhanced in TLR7ko over WT mice. Thus, we show that a virus-sensing PRR modulates the progression of secondary pneumococcal infection following IAV. However, the fatal overall outcome in WT as well as TLR7ko hosts suggests that processes distinct from TLR7-triggering override the contribution of this single PRR.
Introduction
A retrospective study of the 1918/19 Spanish flu has shown that the majority of fatalities were not due to the primary infection with influenza A virus (IAV) but rather severe bacterial superinfections. In these, Streptococcus pneumoniae was the most common bacterial pathogen [1]. This also holds true for later pandemics, including the swine flu outbreak in 2009 [2,3,4]. A conclusive molecular explanation of how a previous IAV infection can leave the host’s antibacterial defense in a paralyzed state for up to several months has not yet been found [5]. However, several mechanisms by which IAV infection modulates the immune system, rendering it incapable of mounting an adequate antibacterial response, have been revealed lately [6,7,8,9]. Yet, by which molecular recognition events this immune modulation is triggered has remained elusive. Clearly, a detailed understanding of the underlying mechanisms will be required to meet the threat of new influenza pandemics in combination with the increasing number of circulating antibiotic-resistant strains of relevant bacterial pathogens.
The innate immune system possesses pattern recognition receptors (PRR) that bind conserved structures of pathogenic microorganisms and trigger defined immune reactions. An extensively studied group of these receptors is the toll-like receptor (TLR) family comprising TLRs 1–11, which are critically linked to the defense against various pathogens [10,11]. The single-stranded negative-sense IAV genome is recognized by TLR7 and 8, which are localized in the endosomal membrane of expressing cells [12,13]. Triggering of TLR7 leads to the release of cytokines and type I IFNs, and can mediate profound local and systemic effects, e.g. the induction of peripheral blood lymphopenia and modulation of immune responses [14,15]. Previous studies in IAV-infected animals have revealed that in the absence of the adaptor protein MyD88, signaling through RIG-I is sufficient for mounting effective initial immune responses and that selective TLR7-deficiency has distinct effects on shaping adaptive immunity [16,17,18,19,20]. Regarding the contribution of TLR7 to the innate responses following IAV infection, increased accumulation of myeloid-derived suppressor cells in the lungs of TLR7ko animals has recently been shown [18]. However, the potential role of TLR7 in bacterial superinfection following influenza has not been addressed so far. While these are most commonly recognized during ongoing IAV infections, other respiratory viruses also predispose for secondary bacterial complications. These include rhinoviruses, respiratory syncytial virus and measles virus [21,22]. Interestingly, most of these viruses possess a single-stranded RNA genome and therefore potentially supply a TLR7-trigger [23,24]. This makes it conceivable that TLR7 triggering during the viral infection mediates some of the immune-modulatory effects which subsequently facilitate bacterial superinfections.
Therefore, in order to clarify how TLR7-mediated anti-IAV responses possibly impact the synergism between respiratory viral and secondary bacterial infections, we have analyzed TLR7-deficient mice during experimental IAV infection and co-infection with S. pneumoniae. As others, we find that the course of IAV infection is not strikingly altered in TLR7 deficient hosts. Even though highly conceivable, we did not find TLR7 to modulate the fatal outcome of bacterial superinfection during influenza. Nevertheless, the course of pneumococcal disease occurring during acute IAV infection was significantly delayed in TLR7-deficient hosts. Therefore, our work shows that TLR7 as a virus-sensing PRR, even though not a major contributor, constitutes one of the many factors involved in the synergism of IAV and S. pneumoniae in mammals.
Materials and Methods
Mice
TLR7ko (TLR7–/–) mice [25], back-crossed at least five generations to the C57BL/6 background, were a gift from S. Bauer (Marburg, Germany) and were bred at the animal facility of the Helmholtz Centre for Infection Research (HZI). Age- and sex-matched littermates were used as WT controls in experiments with five generation back-crossed TLR7ko mice. C57BL/6 mice were purchased from Charles River (Sulzfeld, Germany) or Harlan Winkelmann (Borchen, Germany) and used as controls in experiments with ten generation back-crossed TLR7ko. For luciferase reporter mice, IFN-βΔβ-luc/Δβ-luc males were crossed with TLR7–/– females to obtain TLR7–/yIFN-β+/Δβ-luc males or C57BL/6 females to obtain TLR7+/yIFN-β+/Δβ-luc control males.
All animal experiments were approved by the Niedersächsisches Landesamt für Verbraucherschutz und Lebensmittelsicherheit (file No. 33.42502-006/07) for the HZI and the Swedish animal welfare authority (file No. N254/07) for the Karolinska Institute, and were conducted in conformity with the German animal welfare act, the Swedish animal protection legislation and the European Communities Council Directives 86/609/EEC and 2010/63/EU.
Viral and Bacterial Pathogens
Madin-Darby canine kidney cell-derived IAV PR8/A/34 (H1N1) was obtained as described [26]. S. pneumoniae TIGR4 was grown on blood agar (Columbia agar with 5% sheep blood; BD, San Jose, Calif., USA) at 37°C and 5% CO2. Colonies were inoculated into C+Y (casamino acid and yeast extract) or THY (Todd Hewitt Broth, 1% yeast extract) medium, grown to mid-logarithmic phase and diluted to the appropriate concentrations. For S. pneumoniae, bacterial counts in the inoculum used for mouse infection were confirmed by plating serial dilutions.
Viral and Bacterial Infections
For mouse infections, 8- to 12-week-old mice were anaesthetized by isoflurane inhalation or intraperitoneal injection of ketamine/xylozine in the case of luciferase reporter mice. A volume of 25 µl (IAV) or 20 µl (S. pneumoniae) containing the appropriate concentration of virus or bacteria was administered onto the nostrils. For IAV, the dose lethal to 50% of inoculated C57BL/6 mice (MLD50) was determined as previously reported [26]. For S. pneumoniae, bacteria were diluted in growth medium to 5 × 106 CFU/ml. During all mouse infection experiments, animals were sacrificed when observing overt signs of illness or weight loss of more than 25%.
Lung Viral Load
Lungs were homogenized in TriFastFL reagent (PeqLab). RNA was extracted by addition of 1-Br-3-Cl-Propane (Merck), purified by precipitation and treated with DNase (Ambion) prior to reverse transcription to cDNA (M-MLV Reverse Transcriptase; Invitrogen, Carlsbad, Calif., USA). Quantitative real-time PCR of 100 ng cDNA samples was conducted using the Brilliant SYBR Green QPCR Kit (Stratagene) and primers specific for the IAV nucleoprotein (NP). Quantitative real-time PCR of samples containing known numbers of a plasmid carrying the NP sequence (pVI-PmH5-PR8-NP; provided by G. Sutter, Munich, Germany) was performed to obtain a CT-value/NP copy number standard, which was used to quantify NP copy numbers in the samples.
Quantification of IFN-γ in Bronchoalveolar Lavage
Lungs were flushed with 1 ml PBS and a Luminex-based immunoassay (Millipore) was used according to the manufacturer’s recommendations. Samples were acquired on a LiquiChip 100 Workstation (Qiagen) and data were analyzed using the LiquiChip Analyzer 1.0.
Quantification of Leukocytes in Bronchoalveolar Lavage
Lungs were flushed with 1 ml PBS and cells from bronchoalveolar lavage (BAL) were collected by centrifugation. Erythrocytes were lysed by osmotic shock and viable cells were counted to assess the total number of cells/ml BAL. Fc-γ receptor block was performed through incubation with anti-mouse CD16/CD36 (2.4G2) antibody. Cells were stained for CD11b (M1/70), Gr-1 (RB6–8C5), F4/80 (BM8), CD4 (RM4–5) and CD8 (53–6.7) (BD/eBioscience). For analysis, cells were analyzed for Gr1high/CD11b+ neutrophils following the exclusion of macrophages (F4/80+ cells). For each population, the absolute cell number was calculated from the percent population data from flow cytometric analysis.
Flow Cytometry
Flow-cytometric analyses were performed using a FACS Canto instrument (BD) and FACS Diva (BD), Summit (DAKO) or FlowJo (Tree Star) software.
Assessment of S. Pneumoniae CFU Counts
Blood (5 µl) from the tail vein was diluted in PBS and plated on blood agar plates. Lungs were collected in 1 ml of PBS, homogenized and serial dilutions were plated. CFU were counted after 16 h of incubation at 37°C and 5% CO2.
Histology
Lungs were fixed in 4% paraformaldehyde, followed by paraffin embedding, preparation of 4-µm sections and staining with hematoxylin-eosin. Histological evaluation was performed by an animal pathologist certified by the European College of Veterinary Pathologists in a blinded fashion.
Detection of Luciferase Activity in IFN-β+/Δβ-luc Mice
Mice were injected with R-848 in PBS (50 µg i.v.; Invivogen, San Diego, Calif., USA) or intranasally infected with IAV. Detection of luciferase activity was performed by in vivo imaging or ex vivo quantification in lung homogenates as reported previously [27].
In vivo Phagocytosis Assay
S. pneumoniae TIGR4 was grown on blood agar plates. Bacteria were resuspended in PBS, pelleted by centrifugation and incubated in 0.1 mg/ml fluorescein isothiocyanate (FITC) in 0.1 M NaHCO3 (pH 9) for 1 h. Bacteria were washed and resuspended in PBS to ∼1.25 × 108 CFU/ml and used for intranasal inoculation of anaesthetized mice (20 µl). Animals were sacrificed 2 h later and lungs were flushed twice with a total volume of 2 ml PBS. Cells from BAL were collected by centrifugation, erythrocyte lysis and Fc-γ receptor block were performed and cells were stained for F4/80 followed by flow-cytometric analysis.
Statistical Analysis
Statistics were performed by the indicated t tests and survival data were compared by Kaplan Meier analysis log-rank test using Graph Pad Prism 5.00 (Graph Pad Software, La Jolla, Calif., USA).
Results
Increased Body-Weight Loss but Otherwise Unaltered Course of IAV Infection under TLR7 Deficiency
In order to address the response to secondary bacterial infection after sublethal IAV infection in the absence of TLR7, we first characterized survival and weight loss of TLR7ko mice after the viral infection alone. We chose the viral strain A/PR8/34 (H1N1) as an extensively studied IAV representative. Following IAV infection of wild-type (WT) and TLR7ko mice with 0.04 LD50 PR8 virus, no significant differences in survival were detectable (fig. 1a). To address a possible effect of TLR7 present only during infection with higher viral doses, survival following infection with 1.0 LD50 PR8 virus was assessed in both mouse strains. Also here, no effect of TLR7-deficiency became apparent (fig. 1a).
![Fig. 1. TLR7 deficiency leaves survival and viral clearance following IAV infection unaffected but exacerbates body weight loss. a WT and TLR7ko mice were intranasally infected with 0.04 LD50 or 1.0 LD50 influenza PR8 and body weight and survival were assessed over 14 days. b The relative body weight is shown as mean ± SEM (no error bars shown for SEM <1%) of all animals surviving the infection to day 14 [* p = 0.027 (0.04 LD50 day 1) and 0.007 (0.04 LD50 day 2), ** p < 0.001 (1.0 LD50 day 1) as determined by an unpaired, two-sided t test]. c For quantification of airway viral load in WT and TLR7ko mice, lung cDNA was analyzed for influenza NP sequence copy numbers following infection of WT and TLR7ko mice with 0.04 LD50 IAV PR8. Data are shown as mean copy numbers ± SEM (n ≥6/group).](https://karger.silverchair-cdn.com/karger/content_public/journal/jin/5/1/10.1159_000345112/2/m_000345112_f01.gif?Expires=1716324996&Signature=HeaxgWYrX44errt1tQaJZwbyPyb4yk9hmSmOCvLW8L29qLJzK4R8ExoR69qvc1opQCXrmgOQEVBPfTA7PkKnNawvKyxiPQB7zUI7RpM1~rOby9BOJlGDQwV9EN7aSZOlK7p0HcbsElFHHxfrISijRPV2L4DvqWkTEZkFR1-WE3QwAYMruK9343wMGnyMr-RRhGPK0HXAFOcZcxaQDiypAk5iDqGyjR0e6MlhxKhL7MOsqND4LEq3lTw8uJtCPPJVCvA7kAdxQ7SzuAGxE3Ox5SQ4CxwvuOeYZkJboeBI9FT2tAzU4KZ2bVQhi9c0Vq5BFU5d6wdO0gGH90hYVsYuxA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
TLR7 deficiency leaves survival and viral clearance following IAV infection unaffected but exacerbates body weight loss. a WT and TLR7ko mice were intranasally infected with 0.04 LD50 or 1.0 LD50 influenza PR8 and body weight and survival were assessed over 14 days. b The relative body weight is shown as mean ± SEM (no error bars shown for SEM <1%) of all animals surviving the infection to day 14 [* p = 0.027 (0.04 LD50 day 1) and 0.007 (0.04 LD50 day 2), ** p < 0.001 (1.0 LD50 day 1) as determined by an unpaired, two-sided t test]. c For quantification of airway viral load in WT and TLR7ko mice, lung cDNA was analyzed for influenza NP sequence copy numbers following infection of WT and TLR7ko mice with 0.04 LD50 IAV PR8. Data are shown as mean copy numbers ± SEM (n ≥6/group).
TLR7 deficiency leaves survival and viral clearance following IAV infection unaffected but exacerbates body weight loss. a WT and TLR7ko mice were intranasally infected with 0.04 LD50 or 1.0 LD50 influenza PR8 and body weight and survival were assessed over 14 days. b The relative body weight is shown as mean ± SEM (no error bars shown for SEM <1%) of all animals surviving the infection to day 14 [* p = 0.027 (0.04 LD50 day 1) and 0.007 (0.04 LD50 day 2), ** p < 0.001 (1.0 LD50 day 1) as determined by an unpaired, two-sided t test]. c For quantification of airway viral load in WT and TLR7ko mice, lung cDNA was analyzed for influenza NP sequence copy numbers following infection of WT and TLR7ko mice with 0.04 LD50 IAV PR8. Data are shown as mean copy numbers ± SEM (n ≥6/group).
When comparing the body weight loss in the course of IAV infection between both mouse strains, a subtle TLR7-dependent exacerbation was apparent (fig. 1b). We observed increased early weight loss and delayed recovery to the original body weight in TLR7ko animals at both the viral doses tested, which confirms results recently published by Jeisy-Scott et al. [18].
To assess viral clearance in TLR7ko animals, viral NP RNA sequence copy numbers were quantified in the course of infection with 0.04 LD50 IAV (fig. 1c). For these and the following experiments characterizing the anti-IAV response in TLR7ko mice, the lower viral dose was chosen to ensure survival and recovery of nearly all animals within experimental groups. Independent of the presence or absence of TLR7, IAV was cleared with similar kinetics in the lungs of both mouse strains. Taken together, these analyses confirmed previous studies [18,19] that showed that TLR7 is dispensable for survival and viral clearance following respiratory IAV infection.
TLR7 Contributes to the Airway IFN-γ Response but Is Dispensable for the Recruitment of Macrophages and Neutrophils following Respiratory IAV Infection
Respiratory IAV infection leads to a strong induction of cytokines and chemokines in the airways, coordinating function and recruitment of immune cells [28,29,30]. This process is known not to be drastically impaired in TLR7ko hosts [18,19]. Of the inflammatory mediators typically released during IAV infection, IFN-γ was of special interest to our study of the role of TLR7 in secondary bacterial infection. On the one hand, TLR7ko mice have been found to produce less IFN-γ following IAV infection [18] and on the other hand IFN-γ has been shown to be a main contributor in mediating enhanced susceptibility to pneumococci following influenza [7]. Also, in our model of IAV infection, TLR7ko mice revealed impaired IFN-γ production in the airways (fig. 2a). In serum samples obtained throughout the course of the infection, no significant increase in systemic IFN-γ levels could be detected at any of the analyzed time points (data not shown). However, in contrast to previous studies, the inhibition of the airway IFN-γ response was evident only at the early time points in the course of infection, but could still possibly have an impact on IAV-induced macrophage inhibition.

TLR7 contributes to the early airway IFN-γ response but is dispensable for the recruitment of macrophages, neutrophils and T cells following IAV infection. WT (black bars) and TLR7ko (open bars) mice were infected with 0.04 LD50 IAV PR8 and sacrificed at different time points post-infection. a IFN-γ concentrations in BAL were determined and protein levels are shown as means ± SEM of n ≥5 mice/group. The number of cells in BAL was counted and relative contributions of leukocyte subsets assessed by flow-cytometry. Absolute numbers of macrophages, neutrophils (b) as well as CD4+ and CD8+ T cells (c) were calculated from flow-cytometric data and total cell numbers. Data show mean cell counts/ml ± SEM of n ≥6 mice/group and were compared by an unpaired, two-sided t test (* p < 0.005).
TLR7 contributes to the early airway IFN-γ response but is dispensable for the recruitment of macrophages, neutrophils and T cells following IAV infection. WT (black bars) and TLR7ko (open bars) mice were infected with 0.04 LD50 IAV PR8 and sacrificed at different time points post-infection. a IFN-γ concentrations in BAL were determined and protein levels are shown as means ± SEM of n ≥5 mice/group. The number of cells in BAL was counted and relative contributions of leukocyte subsets assessed by flow-cytometry. Absolute numbers of macrophages, neutrophils (b) as well as CD4+ and CD8+ T cells (c) were calculated from flow-cytometric data and total cell numbers. Data show mean cell counts/ml ± SEM of n ≥6 mice/group and were compared by an unpaired, two-sided t test (* p < 0.005).
As the levels of major chemo-attractants released following IAV infection were not altered by TLR7-deficiency (data not shown) [18], it was an expected finding that also the numbers of macrophages and neutrophils (fig. 2b) in the bronchoalveolar lavage of WT and TLR7 hosts were not significantly altered during the course of infection. Since T cells recruited to the lungs in the course of IAV infection are the major source of airway IFN-γ [7], T cell numbers present in bronchoalveolar lavage were assessed over the course of the infection in both WT and TLR7ko mice. CD4+ and CD8+ T cell numbers present in BAL increased over time and there were no significant changes in TLR7ko mice compared to WT (fig. 2c). In addition, T cells isolated from lungs of both mouse strains after infection with PR8 virus showed no difference in in vitro IFN-γ production in response to unspecific stimulation (data not shown). This suggests that cellular sources other than T cells are responsible for the delayed airway IFN-γ response in TLR7-deficient hosts. Taken together, we found TLR7 to significantly contribute to the early IFN-γ production in the respiratory tract but to be dispensable for potent airway anti-influenza responses such as immune cell recruitment.
TLR7 Deficiency Delays Progression of Pneumococcal Superinfection following IAV Infection
While the overall outcome of the viral infection was similar in the absence of TLR7, the early lack of IFN-γ could possibly influence the anti-bacterial defense in situations of bacterial superinfection, as this cytokine has been found to mediate the functional suppression of alveolar macrophages (AM) [7], which are key cells for anti-bacterial defense. To test this assumption, WT and TLR7-deficient mice were superinfected with S. pneumoniae TIGR4 on day 7 following IAV infection, according to a previously established co-infection model [26]. In this model, mice are infected with a low dose of IAV PR8 prior to infection with a low dose of S. pneumoniae. We used 0.04 LD50 of the virus to be able to clearly distinguish between death through the viral infection, which hardly occurs at this dose (fig. 1a), and death through the secondary bacterial infection. In human patients the primary viral and secondary bacterial infection are also reported to be distinguishable. Typically, patients have nearly recovered from the influenza infection when infection with the secondary bacterial pathogen causes a recurrence of symptoms [31]. When infected with S. pneumoniae 7 days after IAV pre-infection, both WT and TLR7ko mice showed profoundly increased mortality compared to bacterial infection alone without a significant difference between the two mouse strains (fig. 3a). Importantly, survival following S. pneumoniae single infection did not significantly differ between WT and TLR7ko mice (fig. 3a). In the chosen model of pneumococcal infection, death due to bacterial infection is accompanied by the systemic spread of bacteria from the lungs via the bloodstream [32]. Therefore, we assessed the mean time point of the onset of bacteremia after bacterial co-infection as a measure of disease progression. Interestingly, in TLR7ko mice, bacteremia following bacterial superinfection developed at a significantly later point in time when compared to WT mice (fig. 3b). This implied that systemic pneumococcal spread was more efficiently controlled in the absence of TLR7. To exclude that this effect was independent of the viral infection, survival and onset of bacteremia were compared between WT and TLR7ko mice following infection with a higher dose of S. pneumoniae alone (online suppl. fig. 1; for all online suppl. material, see www.karger.com?doi=10.1159/000345112). This better resembles the situation in co-infected mice, where high numbers of bacteria are present in the respiratory tract and disseminate through the bloodstream. However, also in the S. pneumoniae single infection with a higher dose, no significant difference in survival (online suppl. fig. 1a) or in the onset of bacteremia (online suppl. fig. 1b) was detected between the two mouse strains, suggesting that the results in co-infected mice can be attributed to the underlying IAV infection. In addition to the survival studies in co-infected animals, the lungs and blood of infected WT and TLR7ko mice were harvested at 4 and 24 h following the bacterial inoculation in order to quantify bacterial CFU. Again, no difference became apparent when comparing WT and TLR7-deficient hosts in the bacterial infection alone (fig. 3c, d). As previously reported and in contrast to naïve animals, IAV-pre-infected WT mice were unable to control bacterial growth in the lower respiratory tract as well as dissemination through the bloodstream [26]. In TLR7ko mice the overall situation was similar. However, co-infected TLR7ko mice, in comparison with co-infected WT mice, carried 55-fold lower mean bacterial counts in lung tissue 24 h following pneumococcal infection (fig. 3c). Even though this difference did not reach the level of statistical significance, it was in line with our finding that TLR7 deficiency delayed disease progression and systemic bacterial spread following pneumococcal infection 7 days after IAV infection. This was further supported by the data from blood cultures (fig. 3d) where at 4 and 24 h following pneumococcal co-infection a higher proportion of WT mice [1/7 (14%) and 4/7 (57%), respectively] carried bacteria in their blood when compared to TLR7ko animals [0/9 (0%) and 2/9 (22%), respectively]. Histopathology of the lungs of co-infected WT and TLR7ko mice revealed a multifocal to confluent chronic bronchointerstitial pneumonia and perivascular and peribronchiolar lymphocytic cuffings – both consistent with the preceding IAV infection – as well as an accompanying suppurative bronchopneumonia indicating the secondary bacterial infection. There were no significant differences between the lungs of WT and TLR7ko mice concerning the quantity of the lesions. However, in co-infected WT mice the quality of the lung lesions was of a more necrotizing character and showed a more conspicuous intra-alveolar fibrin exudation compared to co-infected TLR7ko mice (fig. 3e).

While survival is not affected, progression of secondary bacterial disease following IAV infection is delayed in TLR7-deficient hosts. WT (closed symbols) and TLR7ko (open symbols) mice were intranasally infected with 0.04 LD50 IAV PR8 or left uninfected on day 0 (squares) and infected with 1 × 105 CFU S. pneumoniae on day 7 (circles). a Animals were followed for survival over 7 days after the pneumococcal infection and survival was compared by a log-rank test. b Blood samples were collected at 24, 48 and 72 h and analyzed for the presence of S. pneumoniae CFU. The mean time point for onset of bacteremia (±SEM) was calculated from all positively tested mice and compared by unpaired, two-tailed t tests (p = 0.015). In separate experiments, WT (closed circles) and TLR7ko (open circles) mice were sacrificed 4 or 24 h following the bacterial infection and S. pneumoniae CFU counts in lung homogenates (c) and blood (d) were quantified and compared between mouse strains by unpaired, two-sided t tests. e Histological examination of lungs was performed on uninfected mice at day 7 following IAV infection and 24 h following co-infection. Blood vessels (V), bronchioles (*) and intra-alveolar fibrin exudations (arrows) are indicated. Scale bar = 100 µm. Sections are representative for 2–3 analyzed infected mice per group.
While survival is not affected, progression of secondary bacterial disease following IAV infection is delayed in TLR7-deficient hosts. WT (closed symbols) and TLR7ko (open symbols) mice were intranasally infected with 0.04 LD50 IAV PR8 or left uninfected on day 0 (squares) and infected with 1 × 105 CFU S. pneumoniae on day 7 (circles). a Animals were followed for survival over 7 days after the pneumococcal infection and survival was compared by a log-rank test. b Blood samples were collected at 24, 48 and 72 h and analyzed for the presence of S. pneumoniae CFU. The mean time point for onset of bacteremia (±SEM) was calculated from all positively tested mice and compared by unpaired, two-tailed t tests (p = 0.015). In separate experiments, WT (closed circles) and TLR7ko (open circles) mice were sacrificed 4 or 24 h following the bacterial infection and S. pneumoniae CFU counts in lung homogenates (c) and blood (d) were quantified and compared between mouse strains by unpaired, two-sided t tests. e Histological examination of lungs was performed on uninfected mice at day 7 following IAV infection and 24 h following co-infection. Blood vessels (V), bronchioles (*) and intra-alveolar fibrin exudations (arrows) are indicated. Scale bar = 100 µm. Sections are representative for 2–3 analyzed infected mice per group.
Taken together, these data show that TLR7 triggering during IAV infection contributes to the rapid progression of bacterial disease occurring during acute influenza. However, TLR7 deficiency was clearly not able to rescue mice from the dramatically high post-IAV susceptibility to an otherwise sublethal dose of S. pneumoniae. This highlights the complexity of the exceptional synergism between IAV and bacterial pathogens and the strong impact of additional factors on the underlying mechanisms.
TLR7 Does Not Influence the Outcome of Pneumococcal Superinfection after Recovery from IAV Infection
IAV can induce long-term suppression of antibacterial defense mechanisms, rendering hosts highly susceptible to bacterial superinfection for months after clearance of the primary viral infection [5,8]. Therefore, we also addressed the susceptibility to S. pneumoniae on day 14 following IAV infection. At this time, animals had largely recovered from weight loss and successfully cleared the viral burden. Also here, TLR7ko mice displayed similar survival rates in direct comparison with WT hosts following co-infection on day 14 after IAV (fig. 4).

TLR7 does not influence the outcome of pneumococcal superinfection after recovery from IAV infection. WT (n = 20) and TLR7ko (n = 20) mice were intranasally infected with 0.04 LD50 IAV PR8 14 days before intranasal infection with 1 × 105 CFU S. pneumoniae in at least two independent experiments. Survival was followed over 7 days and compared between mouse strains by a log-rank test. n.s. = Not significant.
TLR7 does not influence the outcome of pneumococcal superinfection after recovery from IAV infection. WT (n = 20) and TLR7ko (n = 20) mice were intranasally infected with 0.04 LD50 IAV PR8 14 days before intranasal infection with 1 × 105 CFU S. pneumoniae in at least two independent experiments. Survival was followed over 7 days and compared between mouse strains by a log-rank test. n.s. = Not significant.
Altered Type I IFN Levels Are Not Responsible for Delayed Progression of Secondary Bacterial Infection in TLR7ko Hosts
IAV infection typically induces a strong release of type I IFNs in the infected mammalian host, triggered through the recognition of viral nucleic acids and replication intermediates by PRRs such as TLR7 and RIG-I [29,33]. The induced type I IFN response has been shown to be a major contributor to the enhanced susceptibility to post-influenza bacterial infections [6]. Even though TLR7 deficient hosts are generally capable of mounting an anti-viral type I IFN response through alternative innate receptors [19], we wanted to address whether altered levels in the airways of TLR7ko mice in the course of IAV infection possibly accounted for the delay in progression of secondary pneumococcal infection. To overcome the drawbacks of other detection systems, such as insensitivity and the need for invasive sample collection [27], and for an improved understanding of the localization and kinetics of this reaction, we aimed at investigating the type I IFN response after respiratory IAV infection in whole animals. To this end, we took advantage of a luciferase-based IFN-β reporter mouse strain [27], which we crossed with TLR7ko mice. The induction of an IFN-β response was analyzed through quantification of luciferase activity in vivo or ex vivo. The functionality of the reporter strains was confirmed by injection of the TLR7-specific ligand R-848, which led to well-detectable luciferase activity exclusively in WT IFN-β+/Δβ-luc reporter mice (online suppl. fig. S1). However, the in vivo analysis of luciferase activity in IAV infected WT and TLR7ko reporter mice yielded only weak signals, which were hardly sufficient for comparing the two mouse strains (fig. 5a). Thus, to allow accurate quantification of the reporter, luciferase activity was analyzed ex vivo. To this end, lungs were harvested 4 and 7 days after IAV infection. The analysis of lung homogenates confirmed that TLR7ko hosts were able to react to IAV infection by inducing IFN-β locally in the lung (fig. 5b). This response was evident at the same time points and reached similar extents as that of WT hosts. Therefore, an altered local type I IFN response by TLR7ko mice in reaction to IAV infection most likely does not account for the alterations found in the progression of secondary pneumococcal infection.
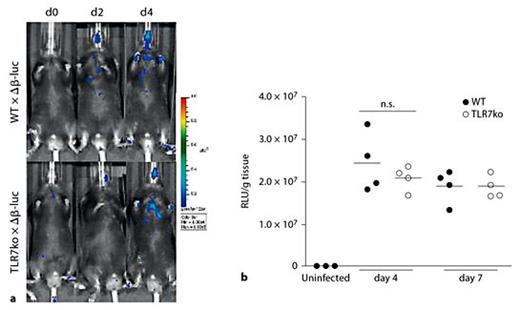
a Absence of TLR7 does not influence the lung IFN-β response following IAV infection. WT and TLR7ko IFN-β+/Δβ-luc reporter mice were intranasally infected with 0.04 LD50 IAV PR8 and induction of luciferase activity was assessed in vivo. One representative of n ≥3 tested animals per mouse strain is shown. b WT (closed circles) and TLR7ko (open circles) IFN-β+/Δβ-luc reporter mice were sacrificed at the indicated time points following IAV infection. Luciferase activity in lungs was quantified ex vivo and is shown as relative light units (RLU)/g tissue for individual mice from one representative out of two experiments. Data were compared by an unpaired, two-sided t test.
a Absence of TLR7 does not influence the lung IFN-β response following IAV infection. WT and TLR7ko IFN-β+/Δβ-luc reporter mice were intranasally infected with 0.04 LD50 IAV PR8 and induction of luciferase activity was assessed in vivo. One representative of n ≥3 tested animals per mouse strain is shown. b WT (closed circles) and TLR7ko (open circles) IFN-β+/Δβ-luc reporter mice were sacrificed at the indicated time points following IAV infection. Luciferase activity in lungs was quantified ex vivo and is shown as relative light units (RLU)/g tissue for individual mice from one representative out of two experiments. Data were compared by an unpaired, two-sided t test.
IAV-Induced Suppression of Macrophage Antibacterial Function Is Alleviated in TLR7-Deficient Hosts
IAV infection has previously been shown to desensitize AM to bacterial TLR-ligands. This in turn led to an inhibition of neutrophil recruitment in response to a secondary bacterial challenge [5]. In order to assess whether this also held true for our co-infection model and whether TLR7 played a role in this mechanism, we characterized the recruitment of effector cells to the airways following pneumococcal infection in naïve mice and mice pre-infected with IAV. In contrast to Didierlaurent et al. [5] we found the number of cells present in BAL 20 h following pneumococcal infection on day 7 after influenza to be strongly and significantly increased compared to that after S. pneumoniae single infection (fig. 6a). The total cell numbers as well as the numbers of neutrophils and macrophages recruited to the airways did not differ between WT and TLR7ko mice, irrespective of the presence and timing of IAV pre-infection (fig. 6a). Thus, an altered recruitment of effector cells following co-infection could not underlie the differences in disease course we found in TLR7-deficient hosts.

Suppression of macrophage function during IAV infection is independent of TLR7 but alleviated in TLR7ko hosts. a WT (solid bars) and TLR7ko (open bars) mice were infected with 1 × 105 CFU S. pneumoniae without previous IAV infection or on day 7 or 14 after infection with 0.04 LD50 IAV PR8 and were sacrificed 20 h later. The total number of cells present in bronchoalveolar lavage was determined and the numbers of macrophages and neutrophils were quantified by flow-cytometry. Data show results of one representative out of two experiments with n ≥3 mice/group. b An in vivo phagocytosis assay was performed to assess phagocytic activity of AM without preceding IAV infection and 7 or 14 days following PR8 infection. Flow-cytometric analysis (c) was performed to quantify the proportion of FITC+/F4/80+ cells out of all F4/80+ macrophages (d) and the respective FITC mean fluorescence intensity (MFI) (e). Groups were compared by an unpaired, two-sided t test. f The ratio of TLR7ko over WT FITC MFI of FITC+ AM was assessed by dividing the mean MFI value of the TLR7ko groups by the value of the corresponding WT group. n.s. = Not significant.
Suppression of macrophage function during IAV infection is independent of TLR7 but alleviated in TLR7ko hosts. a WT (solid bars) and TLR7ko (open bars) mice were infected with 1 × 105 CFU S. pneumoniae without previous IAV infection or on day 7 or 14 after infection with 0.04 LD50 IAV PR8 and were sacrificed 20 h later. The total number of cells present in bronchoalveolar lavage was determined and the numbers of macrophages and neutrophils were quantified by flow-cytometry. Data show results of one representative out of two experiments with n ≥3 mice/group. b An in vivo phagocytosis assay was performed to assess phagocytic activity of AM without preceding IAV infection and 7 or 14 days following PR8 infection. Flow-cytometric analysis (c) was performed to quantify the proportion of FITC+/F4/80+ cells out of all F4/80+ macrophages (d) and the respective FITC mean fluorescence intensity (MFI) (e). Groups were compared by an unpaired, two-sided t test. f The ratio of TLR7ko over WT FITC MFI of FITC+ AM was assessed by dividing the mean MFI value of the TLR7ko groups by the value of the corresponding WT group. n.s. = Not significant.
IFN-γ produced during the anti-IAV response decreases AM scavenger receptor expression and inhibits AM phagocytic activity in secondary bacterial infection [7]. To test whether such a phenomenon was responsible for the effects seen in TLR7ko mice, we assessed AM function in co-infected WT and TLR7ko mice. This was especially interesting as the early airway IFN-γ response after IAV infection was inhibited in TLR7-deficient hosts (fig. 2a). We found that expression of the class A scavenger receptor MARCO was reduced on AM from 7-day IAV-infected WT and TLR7ko mice to the same extent (data not shown). Thus, we also performed in vivo phagocytosis assays in WT and TLR7-deficient mice through intranasal administration of fluorescently (FITC-)labeled S. pneumoniae(fig. 6b). Indeed, AM of 7 days IAV-pre-infected mice displayed a significantly reduced ability to phagocytose S. pneumoniae in the lung. This was mirrored by a reduced frequency of FITC+ macrophages isolated from these animals (fig. 6c, d). This was the case for both WT and TLR7ko mice to the same extent and both mouse strains had partly recovered from this functional suppression by day 14 after the viral infection (fig. 6d). However, when we compared the FITC mean fluorescence intensity as a quantitative measure of the amount of bacteria that macrophages had bound or taken up, AM isolated from TLR7ko animals, irrespective of whether they had been IAV infected 1 or 2 weeks before, displayed significantly higher values after encounter of FITC-labeled S. pneumoniae than those from WT mice (day 7 p = 0.008, day 14 p = 0.0476; fig. 6e). In contrast, the mean fluorescence intensities that were obtained when the assay was performed in uninfected WT and TLR7ko mice were comparable between both mouse strains (fig. 6f). Taken together, these results demonstrated that the overall AM phagocytic capacity was inhibited 7 days following IAV infection irrespective of the presence of TLR7. However, AM from TLR7-deficient hosts were less strongly inhibited in binding and phagocytosis of S. pneumoniae both 7 and 14 days after IAV infection. Even though not sufficient to protect animals from enhanced susceptibility to bacterial superinfection during acute IAV, this possibly at least in part explains the delayed progression of secondary bacterial disease during acute influenza which we observed in TLR7ko hosts.
Discussion
In this study we have addressed the role of TLR7 in the immune response towards bacterial superinfection. In our mouse model of IAV infection alone, TLR7 deficiency neither had an impact on the outcome of the infection nor on clearance of the virus. This is in line with findings from a study by Koyama et al. [19], where signaling by the PRR RIG-I was found to compensate for TLR7 deficiency in the initial response to IAV infection. As recognition of one pattern by different receptors offers additional opportunities for orchestrating the triggered responses, it is highly conceivable that TLR7 plays a role in fine-tuning initial responses, as shown for IFN-γ production, and adaptive anti-IAV immunity [16,17,18,19], rather than constituting a receptor which is responsible for the induction of an effective defense alone.
Next to the resolution of the viral infection as such, protection against secondary bacterial pathogens plays a crucial role in IAV infections. This is implied by the high incidence of severe disease and deaths through secondary infections in IAV-infected patients as observed for past and present pandemics. During recent years, various studies have addressed this synergism between viral and bacterial pathogens and have identified several ways by which viral infections modulate the defense against secondary bacterial infections [5,6,7,8,9,34,35,36,37,38]. The detailed underlying mechanisms, however, still remain incompletely understood. Up to now, studies addressing the role of PRRs in conferring enhanced susceptibility to bacterial superinfection after influenza have selectively addressed the role of bacteria-sensing receptors. TLR2 has been shown not to play a role in the induction of this phenomenon directly [39], whereas others demonstrated that IAV infection leads to an impressively long-lasting general desensitization of the bacteria-recognizing TLR 2, 4 and 5. This in consequence facilitates secondary bacterial infections [5]. However, to our knowledge it has not been considered that also a virus-sensing PRR might affect host responses towards subsequent bacterial infections. A distinct role of TLR7 becomes especially conceivable when taking into account that most viruses predisposing for bacterial superinfection carry a single-stranded RNA genome, thus providing a common innate stimulus [21,22]. Supporting such a concept, we show here that a virally triggered PRR indeed affects the defense against a secondary bacterial pathogen. While TLR7 deficiency does not drastically impact the effectiveness of anti-IAV responses, it delayed the progression of a secondary pneumococcal infection. Ultimately however, this failed to protect from high mortality to low-dose S. pneumoniae infection following IAV, thereby highlighting that the influence of TLR7-dependent factors in this synergism is only little and most likely inferior to other mechanisms playing a role.
Type I IFNs can modulate host immunity in several ways [6,40] and distinct aspects of this modulation are achieved also through selective triggering of TLR7 by synthetic ligands [14,26]. During in vivo IAV infection, the induction and kinetics of the overall type I IFN response are not exclusively mediated by TLR7, but also through triggering of additional innate immune receptors (fig. 5) [19,33]. Therefore, the presence of high levels of type I IFNs following IAV infection also in TLR7-deficient hosts could at least in part explain why the demonstrated contribution of TLR7 triggering to enhanced susceptibility to bacterial superinfection is not able to protect from the fatal outcome.
Another mechanism by which influenza virus infection has been shown to mediate enhanced susceptibility to bacterial superinfection is by modulating the effectivity of antibacterial functions of AM. A reduced capacity of these cells to produce chemokines and recruit neutrophils in response to bacterial TLR-triggering and compromised antibacterial phagocytic functions have both been shown to be consequences of influenza virus infection [5,7]. IFN-γ produced during the viral infection was found to mediate the suppression of AM phagocytosis during secondary bacterial infection, which in turn led to uncontrolled outgrowth of the bacteria in pre-infected animals [7]. Our findings suggest that TLR7 deficiency has an impact on this IAV-induced suppression of macrophage function as we found AM from IAV-infected TLR7ko mice to bind and take up S. pneumoniae more efficiently both during acute viral infection and after its resolution. AM have been found to be crucial for ensuring sterility of the lower respiratory tract and they are key players in the early defense against S. pneumoniae infection [41]. They fulfill this role by the binding, uptake and killing of bacteria. The disadvantage we found for AM of IAV-pre-infected WT compared to TLR7ko mice in binding and phagocytosing pneumococci in the respiratory tract might directly or indirectly have led to the higher bacterial numbers found in the lungs of WT mice 24 h following co-infection on day 7 after IAV infection. In a second step of the anti-bacterial defense neutrophils are recruited to the respiratory tract, a process we found to be unaltered when comparing co-infected mice of both strains. However, the strong bacterial outgrowth in the lungs of co-infected animals apparently overwhelmed the recruited cells which were not able to prevent further spread of the bacteria in either WT or TLR7-deficient hosts. It is possible that, even though the lung bacterial burden did not differ significantly between the two mouse strains, the critical threshold of lung bacterial outgrowth, at which bacteria start to spread from the respiratory tract throughout the organism via the bloodstream, was reached at a significantly earlier time-point in co-infected WT rather than TLR7ko mice. Therefore, in the absence of TLR7, a less severe inhibition of AM following IAV infection possibly accounts for the reduced bacterial burden found in the lungs of co-infected mice and the significantly delayed systemic dissemination of bacteria as well as the absence of long-term immune suppression.
Even though the effects of the virus-sensing PRR TLR7 on post-influenza anti-bacterial defense are slight and a lack of TLR7 did not lead to enhanced survival in bacterial superinfection, our study adds a new aspect to the increasing understanding of the mechanisms underlying the synergism between IAV and S. pneumoniae. However, further studies will be needed to identify the routes through which TLR7 triggering by IAV influences the rapid progression of secondary bacterial disease. Clearly, a detailed knowledge of all of the involved mechanisms as well as the relative importance of their impact will be necessary to face this dangerous threat to human health and to be able to exploit all possible aspects for future strategies of prevention and treatment, possibly including the option of modulating PRR-signaling. This is well demonstrated by studies regarding TLR2, which, even though previously shown not to influence susceptibility to severe secondary bacterial disease after influenza [39], was later found to actually play an important role in mediating immunopathology during its treatment [42].
Acknowledgements
We thank H. Frauendorf (Immuneregulation, Helmholtz Centre for Infection Research, Braunschweig) for the preparation of the pVI-PmH5-PR8-NP plasmid standard.
This work was supported by the International Research Training Group 1273 and by the grant BR2221/1-1/GU769/5-1, both funded by the German Research Foundation (DFG). D.B. is supported by the President’s Initiative and Networking Fund of the Helmholtz Association of German Research Centers (HGF) under contract No. W2/W3-029.
References
D. Bruder and M. Gunzer contributed equally to the study.
