

Abstract
Hyperinsulinaemic hypoglycaemia is a cause of persistent hypoglycaemia in the neonatal and infancy periods. Prompt recognition and management of patients with hyperinsulinaemic hypoglycaemia are essential, if brain damage and long-term neurological sequelae are to be avoided. Hyperinsulinaemic hypoglycaemia can be transient, prolonged, or persistent (congenital). Advances in the fields of molecular biology, genetics, and pancreatic β-cell physiology are beginning to provide novel insights into the mechanisms causing congenital forms of hyperinsulinism. So far mutations in six different genes have been described that lead to unregulated insulin secretion. The histological differentiation of focal and diffuse congenital hyperinsulinism has radically changed the surgical approach to this disease. Until recently, highly invasive investigations were performed to localize the focal lesion, but recent experience with 18F-L-dopa positron emission tomography scanning suggests that this technique is highly sensitive for differentiating diffuse from focal disease as well as for accurately locating the focal lesion. Despite recent advances, the genetic basis of congenital hyperinsulinism is still unknown in about 50% of the patients, and the management of medically unresponsive diffuse disease remains a real challenge.
Introduction
Hyperinsulinaemic hypoglycaemia (HH) is characterized by the unregulated secretion of insulin from pancreatic β-cells in relation to the blood glucose concentration. HH is the commonest cause of persistent hypoglycaemia in the neonatal and infancy periods. As HH is a major risk factor for brain damage and subsequent neurodevelopment handicap [1,] identification and prompt management of patients with HH are essential, if brain damage is to be avoided.
HH can be either transient or persistent (congenital hyperinsulinism; CHI). Transient forms of HH are usually secondary to conditions such as maternal diabetes mellitus or intra-uterine growth retardation [2]. In contrast, recent advances in cellular physiology and molecular biology have begun to unravel the complex mechanisms that lead to congenital forms of hyperinsulinism. Pathologically, CHI can be classified into two major subgroups: ‘channelopathies’ and ‘metabolopathies’ [3]. Channelopathies refer to defects in the pancreatic β-cell ATP-sensitive potassium channels (KATP channels) that lead to unregulated insulin secretion. Metabolopathies cause CHI either by altering the concentration of intracellular signaling molecules (such as ATP/ADP) or by accumulation of intermediary metabolites.
The histological differentiation of CHI into focal and diffuse disease has radically changed the surgical management of patients with CHI [4]. Correct localization and limited excision of the focal lesion will result in complete cure of the patient. Recent advances in 18F-L-dopa PET (positron emission tomography) scanning are beginning to provide greater accuracy in pre-operative differentiation of focal and diffuse disease and in correct localization of focal lesions [5]. In contrast, medically unresponsive diffuse disease will still require a near-total pancreatectomy, greatly increasing the risk of post-pancreatectomy diabetes mellitus.
HH is also observed in several overgrowth syndromes such as Beckwith-Wiedemann and Sotos’ syndromes [6, 7], in association with rare metabolic conditions (congenital disorders of glycosylation; CDG) [8,] and several novel causes of postprandial HH have been recently reported [9, 10]. Table 1 summarizes the different causes of HH.

The aim of this mini review is to outline the clinical presentation, the diagnostic cascade, the pathophysiology, and the management of HH with a particular focus on CHI. Recent reviews focusing on the molecular mechanisms of HH have been published elsewhere [11].
Congenital Hyperinsulinism
Overview
CHI is an extremely heterogeneous disorder with respect to clinical presentation, pancreatic histology, and molecular biology. Both sporadic and familial variants of CHI are recognized, with sporadic forms being relatively uncommon (incidence 1/40,000 live births) and familial forms being common in communities with high rates of consanguinity; in these communities, the incidence may be as high as 1/2,500 live births [12]. The clinical severity of CHI varies mainly with age at onset of hypoglycaemia (severe hypoglycaemia in neonates) and has major consequences in terms of therapeutic outcome and genetic counseling.
Clinical Presentation and Diagnosis
CHI typically presents in the first few days after birth in term and preterm infants with symptomatic hypoglycaemia [13]. The patients may present with non-specific symptoms of hypoglycaemia such as poor feeding, lethargy, and irritability or symptoms such as seizures and coma. Subtle forms of CHI, however, may present later in infancy or even childhood. The hypoglycaemia is usually persistent, and normoglycaemia can only be achieved by giving the infant concentrated intravenous dextrose infusions. Some infants with CHI are macrosomic which may reflect their exposure to perinatal hyperinsulinaemia, but the absence of macrosomia does not exclude CHI, as not all infants with CHI are macrosomic. Some patients with CHI may have mild facial dysmorphism such as a high forehead, a small nasal tip, and short columella with a square face, although the reason for this is unclear [14].
In CHI a blood sample taken at the time of hypoglycaemia will show an inappropriately raised serum insulin level with low serum fatty acid levels and ketone bodies [15]. The low serum fatty acid levels and ketone bodies reflect the metabolic ‘footprint’ of insulin action. Unregulated insulin secretion increases the glucose consumption by insulin-sensitive tissues, such as muscle, adipose tissue, and liver, while simultaneously suppressing hepatic glucose production (both glycogenolysis and gluconeogenesis), lipolysis, and ketogenesis. Due to this metabolic ‘footprint’ of insulin action, the intravenous glucose infusion rate required to maintain normoglycaemia is increased (>8 mg/kg/min, normal 4–6 mg/kg/min). There is no correlation between the serum insulin level and the severity of hypoglycaemia. A ‘normal’ insulin level for normoglycaemia is usually inappropriate in the presence of hypoglycaemia, especially taken in the context of a high glucose requirement to maintain normoglycaemia [15].
The serum lactate level may be elevated in some forms of HH [16,] and the serum ammonia concentration must be measured in all patients presenting with HH because of the association with hyperinsulinism/hyperammoniaemia (HI/HA) syndrome [17]. Urinary organic acid and acylcarnitine analysis should also be performed, since short-chain L-3-hydroxyacyl-CoA dehydrogenase (SCHAD) deficiency can cause CHI [18]. The serum cortisol and glucagon counterregulatory hormonal responses may be blunted in CHI, but replacement therapy with glucocorticoids does not seem to affect the severity of the disease [19, 20].
Some infants and children present with more subtle forms of CHI and require further investigations to aid in the diagnosis. In these patients other supportive evidence, such as decreased serum insulin-like growth factor binding protein-1 levels (as insulin suppresses the transcription of the IGFBP1 gene) [21] and a positive glycaemic response to intramuscular/intravenous glucagon at the time of hypoglycaemia [22,] will provide diagnostic clues. Provocation testing (such as protein or leucine loading and exercise test) will exacerbate the hypoglycaemia in those patients with protein/leucine sensitivity and exercise-induced hypoglycaemia [17, 23]. Figure 1 outlines the diagnostic and management cascade for patients with CHI.

Transient HH
Transient HH is a poorly defined term, as there is no definition of the precise duration of the hypoglycaemia. Classically transient HH is observed in newborns with intra-uterine growth retardation, in those suffering from perinatal asphyxia, in those born to insulin-dependent and gestational diabetic mothers, and in infants with Rh isoimmunization [24]. In these conditions, HH is usually a transient phenomenon and settles within a few days after delivery. However, some neonates (both intra-uterine growth retarded and appropriate weight for gestational age groups) can have prolonged HH that requires treatment with diazoxide, persists for several months, and then resolves spontaneously [25, 26].
Syndromic Forms of HH
Table 1 lists the syndromes which have been reported in association with HH [6, 7,27,28,29,30,31,32]. The mechanism/s of HH in most of these syndromes is/are not known. Beckwith-Wiedemann syndrome is the most common syndrome associated with HH. Beckwith-Wiedemann syndrome is characterized by prenatal and/or postnatal overgrowth, macroglossia, anterior abdominal wall defects, organomegaly, hemihypertrophy, ear lobe creases, helical pits, and renal tract abnormalities. The incidence of HH in children with Beckwith-Wiedemann syndrome is about 50%. This hypoglycemia can be transient, which, in the majority of infants, will be asymptomatic and resolve within the first few days of life. In about 5% of these children, the HH can be persistent and extend beyond the neonatal period, requiring either continuous feeding, medical therapy, or, in rare cases, partial pancreatectomy.
Metabolic Conditions Associated with HH
HH has been described in CDG (Congenital Disorders of Glycosylation), mostly in CDG-Ib but also as the leading symptom in CDG-Ia [8, 33]. Persistent HH as the leading symptom has recently been reported in CDG-Id associated with islet hyperplasia on postmortem examination of the pancreas [34]. Hence if CDG syndrome is suspected, a transferrin isoelectric focusing pattern should be requested. Patients with tyrosinaemia type I may also have HH with islet cell hyperplasia which is not due to impaired insulin clearance, but possibly related to the accumulation of toxic metabolites [35]. In patients with tyrosinaemia type I, urinary organic acids will show excretion of succinylacetone.
Postprandial HH
Several novel syndromes causing postprandial HH have recently been reported (table 1). A syndrome of autosomal dominant postprandial HH with onset in adolescence to adulthood and linked to a mutation (Arg1174Gln) in the insulin receptor kinase has been reported [9]. In these patients a prolonged (5 h) oral glucose tolerance test demonstrates marked postprandial HH, with clamp studies showing reduced insulin sensitivity and clearance of serum insulin in affected family members as compared with control subjects. In adults, a syndrome of ‘noninsulinoma pancreatogenous’ hypoglycaemia has been recognized [10]. These patients demonstrate neuroglycopaenic episodes from HH within 4 h of meal ingestion and have negative 72-hour fasts.
Pathophysiology of HH
Transient HH
Aetiology and the mechanisms responsible for transient forms of HH are unclear. Although the aetiology of these transient forms of HH is not thought to be genetic, recently mutations in the gene encoding the hepatic nuclear transcription factor-4α (HNF4A gene) have been reported [36]. In these patients the birth weight of the heterozygote HNF4A mutation carriers was dramatically increased. However, it is not known how mutations in HNF4A gene cause transient HH.
Congenital HH
CHI can be classified into ‘channelopathies’, where defects in the pancreatic β-cell KATP channels lead to unregulated insulin secretion, or ‘metabolopathies’, with increased β-cell ATP formation or accumulation of intermediary metabolites, triggering insulin secretion.
CHI due to Channelopathies
The commonest genetic causes of CHI are autosomal recessive mutations in the genes ABCC8 and KCNJ11 (encoding the two subunits SUR1 and KIR6.2, respectively) of the pancreatic KATP channels [37, 38]. Autosomal dominant mutations have also been described [39]. KATP channels play a pivotal role in transducing metabolic signals to electrical changes in membrane potential (fig. 2). These mutations result in differing abnormalities of recombinant KATP channels, including protein folding, protein synthesis defects, assembly and trafficking defects, and alterations in both nucleotide regulation and open-state frequency [40,41,42,43].
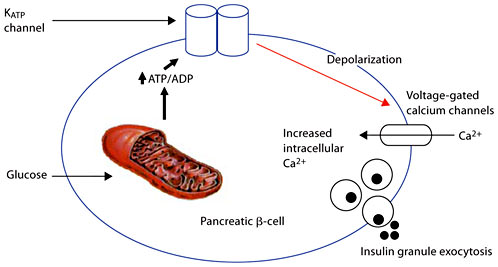
Role of KATP channels in linking glucose metabolism to regulated insulin. KATP channels play a pivotal role in transducing metabolic signals to electrical changes in membrane potential. The metabolism of glucose in the β-cell increases the ratio of ATP/ADP which has the effect of closing the KATP channels. This in turn causes the opening up of voltage-gated calcium channels which regulate the entry of calcium into the β-cell. The entry of calcium is thought to be the final stimulus for insulin exocytosis.
Role of KATP channels in linking glucose metabolism to regulated insulin. KATP channels play a pivotal role in transducing metabolic signals to electrical changes in membrane potential. The metabolism of glucose in the β-cell increases the ratio of ATP/ADP which has the effect of closing the KATP channels. This in turn causes the opening up of voltage-gated calcium channels which regulate the entry of calcium into the β-cell. The entry of calcium is thought to be the final stimulus for insulin exocytosis.
Mutations that affect the regulation of the KATP channels by altering their sensitivity to changes in ADP/ATP will also lead to an unregulated insulin secretion. Several mutations have now been described that result in the loss of ADP-dependent gating properties of the channel [39, 44, 45]. Loss of ADP-dependent gating results in the constitutive inhibition of KATP channels by ATP.
In more than 50% of the patients, screening has failed to define the genetic basis of CHI. There are families with autosomal dominantly inherited HH, but no underlying genetic defects have been found [46]. In some populations, mutations in the ABCC8 gene account for only about 20% of the cases of CHI [47,] suggesting that other genes may be involved.
CHI due to Metabolopathies
Glutamate Dehydrogenase (GDH). Activating mutations in GDH are the second commonest cause of CHI. Activating mutations in GDH underlie the molecular basis of the HI/HA syndrome and may explain the ‘leucine-sensitive’ hypoglycaemia described in previous years [17]. The HI/HA syndrome is caused by missense mutations of GDH that reduce the sensitivity of the enzyme to allosteric inhibition by the high-energy phosphates GTP and ATP. GDH is allosterically activated by leucine and inhibited by GTP [48]. Mutations which cause loss of inhibition by GTP cause leucine to increase the oxidation of glutamate, thereby raising the ratio of ATP/ADP in the pancreatic β-cell. The increased ratio of ATP/ADP then triggers closure of the KATP channel, opening the voltage-gated calcium channel, raising cytosolic calcium, and triggering the release of insulin.
Patients with the HI/HA syndrome can present with hypoglycaemia either in the neonatal period or later on in childhood. These patients also have a mildly elevated plasma ammonia concentration which appears to be asymptomatic. Patients show no signs of lethargy or headaches, typical of other forms of hyperammonaemia. The mechanism of the hyperammonaemia is still unclear at present. These patients typically demonstrate protein-induced HH (leucine sensitivity), but also have fasting hypoglycaemia.
Children with the HI/HA syndrome have an unusual frequency of absence-type seizures [49]. These children have an EEG pattern of generalized epilepsy that resembles the seizures associated with mutations of plasma membrane ion channels. It is unlikely that this seizure pattern is a manifestation of ammonia toxicity.
Glucokinase (GCK). GCK is the rate-limiting step in the metabolism of glucose and acts as the cellular sensor of glucose concentrations. Activation of GCK lowers the threshold for glucose-stimulated insulin secretion (‘resetting’ of the glucose-stimulated insulin release threshold), thus causing hypoglycaemia. The first activating mutation in the GCK gene was Val455Met which was a single-base change, resulting in the substitution of methionine for valine at codon 455 [50]. When expressed in vitro, the Val455Met mutation increased the affinity of GCK for glucose. Several other patients with activating mutations in the GCK gene have now been reported [51, 52,] all responsive to medical therapy with diazoxide. However a case of severe HH due to a ‘de novo’ mutation in GCK gene (Y214C) was reported which failed to respond to medical therapy [53]. Functional studies of this mutant showed a sixfold increase in its affinity for glucose, and histology of the resected pancreas in this patient revealed abnormally large and hyperfunctional islets. It is unclear, why this patient failed to respond to diazoxide, one possibility being that the dose of diazoxide was insufficient.
SCHAD Deficiency. SCHAD, encoded by the HADHSC gene, is an intramitochondrial enzyme that catalyzes the penultimate reaction in the β-oxidation of fatty acids, the NAD+-dependent dehydrogenation of 3-hydroxyacyl-CoA to the corresponding 3-ketoacyl-CoA. So far 3 patients with mutations in the HADHSC gene and HH have been reported [18, 54, 55]. The clinical presentation can be heterogeneous, either with mild late-onset hypoglycaemia or severe neonatal hypoglycaemia. The acylcarnitine profile in all reported patients has demonstrated raised hydroxybutyrylcarnitine levels, and urinary organic acids showed raised 3-hydroxyglutarate concentrations with decreased expression and function of SCHAD.
The mechanism of how a defect in the HADHSC gene leads to dysregulated insulin secretion is unclear at present. Fatty acids increase insulin secretion by affecting the concentrations of long-chain fatty acyl derivatives as a result of the inhibitory effect of citrate and malonyl-CoA on the rate-controlling carnitine palmitoyltransferase-1, but it is unclear how defects in SCHAD lead to unregulated insulin secretion. Interestingly, Foxa2 (HNF3 β) has recently been shown to be involved in regulating the expression of the HADHSC gene, with studies in Foxa2-deficient β-cells showing a threefold downregulation of HADHSC gene transcripts along with the ability of Foxa2 to bind to and activate this gene [56]. Further studies will give new insights into how defects in the HADHSC gene lead to HH. Figure 3 summarizes the known genetic causes of HH.

Summary of the known genetic causes of HH. Mutations in the genes ABCC8 and KCNJ11 (encoding SUR1 and KIR6.2, respectively, of the KATP channel) are the commonest causes of CHI. The molecular mechanisms leading to HH due to mutations in the genes HADHSC (encoding SCHAD) and HNF4alpha are still unclear. HNF4A = Hepatic nuclear transcription factor 4 alpha; NH3 = ammonia. For explanation of the other abbreviations see text.
Summary of the known genetic causes of HH. Mutations in the genes ABCC8 and KCNJ11 (encoding SUR1 and KIR6.2, respectively, of the KATP channel) are the commonest causes of CHI. The molecular mechanisms leading to HH due to mutations in the genes HADHSC (encoding SCHAD) and HNF4alpha are still unclear. HNF4A = Hepatic nuclear transcription factor 4 alpha; NH3 = ammonia. For explanation of the other abbreviations see text.
Exercise-Induced HH. In exercise-induced HH, strenuous physical exercise leads to inappropriate insulin release from β-cells causing postexercise hypoglycaemia [23]. These patients show an increased insulin secretion in response to intravenous pyruvate administration in comparison with control patients [23]. The molecular mechanism/s of how exogenous pyruvate triggers an inappropriate insulin secretion in these patients is/are still unclear.
Histology of CHI
Nesidioblastosis is a histological term which describes islets budding off from exocrine ducts but it is not specific for CHI [4]. It can also be observed in some normoglycaemic infants. Although two major histological forms of the disease have been described (diffuse and focal), there are still some cases which represent a diagnostic challenge, as they cannot be easily classified into focal or diffuse [57]. Both the diffuse and focal forms share a similar clinical presentation, but result from different pathphysiological and molecular mechanisms. In addition, diffuse CHI usually presents as an autosomal recessive disorder, whereas focal CHI is sporadic.
The typical diffuse form affects all the β-cells and is most commonly due to recessive mutations in the genes encoding the two subunits of the KATP channel. Typical diffuse disease is characterized by an increase in the size of the pancreatic β-cell nuclei throughout the pancreas.
The ‘focal’ form (focal adenomatous pancreatic hyperplasia) of CHI is found in about 40–50% of the children and appears to be localized to one region of the pancreas. The genetic defect in the focal form consists of germline mutations in the paternal allele of ABCC8 and KCNJ11 genes, encoding SUR1 and KIR6.2, respectively, on chromosome 11p15. In addition, the lesion exhibits a somatic loss of a part of the maternally inherited chromosome 11p which includes imprinted maternally expressed tumour suppressor genes (H19 and P57KIP2), paternally expressed insulin growth factor-2, as well as (non-imprinted) SUR1/Kir6. This results in a corresponding reduction to homozygosity of the paternal mutation, and the outcome is unregulated insulin secretion. β-Cells within the focal lesion do not express p57KIP2, but insulin growth factor-2 is mildly increased. The somatic loss of heterozygosity is associated with increased proliferation [58, 59]. The focal lesion is different from the insulinoma (also called adenoma) in histology and molecular mechanisms of insulin secretion [60].
General Management
The management of patients with HH can be extremely complicated (as they have multiple problems, such as fluid overload, cardiac failure, and sepsis). They will require frequent blood glucose monitoring and the insertion of a central venous catheter to deliver concentrated dextrose infusions. Ideally, these patients should be referred to specialized centres that have the necessary multidisciplinary team experience and expertise in managing them [15]. The treatment of HH involves medical therapy and surgery in some cases. The mainstay of initial medical treatment is the provision of adequate carbohydrate to maintain normoglycaemia (3.5–6 mmol/l). Adequate carbohydrate can be provided as intravenous glucose at high concentrations, together with a nasogastric feeding tube for regular feeds. Concentrated dextrose infusions should be delivered using a central venous catheter (Hickman line) or an umbilical venous catheter. Glucose polymer can be added to the enteral feed to increase the carbohydrate intake. Some infants may require the insertion of a gastrostomy for regular and frequent feeds.
Medical Management
Table 2 is a summary of the medications used in the treatment of CHI, their doses, side effects, and the possible mechanisms of actions. Diazoxide is the first-line treatment of choice. Diazoxide and nifedipine are given orally, whereas octreotide and glucagon are given subcutaneously or intravenously. Nifedipine is a calcium channel antagonist and has been used in some patients with CHI, although the vast majority of patients fail to show any response. Despite this, there have been several reports of nifedipine-responsive CHI patients [61, 62,] but the underlying molecular pathophysiology of CHI in these reported cases is unclear.

Diazoxide
The clinical effectiveness of diazoxide is variable [63]. Mutations in the ABCC8/KCNJ11 gene are not predictive of the response to diazoxide, and there is no correlation between the histology and the clinical efficacy of diazoxide [64]. Patients with transient and syndromic forms of HH will usually respond to diazoxide, whereas those with severe neonatal CHI will show no response.
A newer more potent synthetic diazoxide analog, 6,7-dichloro-3-isopropylamino-4H-1,2,4-benzothiadiazine 1,1-dioxide (BPDZ 154), has been evaluated [65]. In patients with CHI associated with severe loss of KATP channel function, neither diazoxide nor BPDZ 154 was effective in suppressing insulin release in vivo. In contrast, in those cases of CHI, where the KATP channel function was preserved, BPDZ 154 was able to activate KATP channels in vitro, while diazoxide had no therapeutic effect when used in vivo.
These findings imply that increasing the concentrations of diazoxide in vivo may prove beneficial, but in clinical practice increasing doses of diazoxide are associated with side effects.
Octreotide
Octreotide is a long-acting analog of the natural hormone somatostatin and is used in the short- and long-term management of CHI. In the short term (with and without glucagon), it is used to stabilize patients pending further investigations. Octreotide has been successively used in the long-term management of some CHI patients in combination with frequent feeding [66]. The long-term medical management of diffuse disease with octreotide and frequent feeding should not be taken lightly, as it may impose a huge burden and be stressful on the family. A gastrostomy is recommend in these patients, as this will allow the delivery of bolus and continuous overnight feeds.
Glucagon
Glucagon is used for the acute management of hypoglycemia, when there are adequate glycogen stores (e.g., hyperinsulinism either due to endogenous hyperinsulinemia or to exogenous insulin treatment of patients with diabetes). It is also used in the short term to stabilize patients with HH in combination with octreotide. At higher doses (>20 µg/kg/h), glucagon is a potent stimulator of insulin secretion [67].
Differentiating Focal from Diffuse CHI
Identification of those children, who have the focal form of the disease preoperatively, is a critical part of the management of patients with CHI. The preoperative localization allows radically different treatment options and medical outcomes. Focal disease is curable with limited (partial) pancreatectomy with few long-term complications. Until recently, highly invasive methods such as intrahepatic pancreatic portal venous sampling, the intra-arterial calcium stimulation/venous sampling test, and acute insulin response testing to intravenous glucose, calcium, and tolbutamide were used for identifying those children with focal and diffuse forms of the disease. More recently, 18F-L-dopa PET has been successfully used to localize the focal domain [5, 68]. The principle of this test is based on the fact that islets take up L-dopa and convert it to dopamine by dopa decarboxylase which is present in the islet cells [69]. 18F-L-dopa PET can also accurately locate ectopic focal lesions [70]. In figure 4 the 18F-L-Dopa PET/CT scan shows a focal lesion embedded in the head of the pancreas.

18F-L-dopa PET/CT scan showing a focal lesion embedded in the head of the pancreas. For comparison of tracer uptake in various parts of the pancreas, standardized uptake values (SUVs) were calculated: The 18F-L-dopa uptake is greatest in the head (SUV 5.0) of the pancreas as compared with body and tail (SUV 3.2 and SUV 3.0, respectively).
18F-L-dopa PET/CT scan showing a focal lesion embedded in the head of the pancreas. For comparison of tracer uptake in various parts of the pancreas, standardized uptake values (SUVs) were calculated: The 18F-L-dopa uptake is greatest in the head (SUV 5.0) of the pancreas as compared with body and tail (SUV 3.2 and SUV 3.0, respectively).
Natural History and the Role of Surgery in CHI
The role of surgery in focal CHI is relatively well defined. If a focal lesion is identified and accurately located, it should be surgically removed, as this will ‘cure’ the patient. There is early experience, showing that some focal lesions may be removed laparoscopically [71].
As diffuse CHI is a heterogeneous disorder with respect to clinical presentation and response to medical therapy, the role of surgery in those cases that are diazoxide unresponsive is not so clear. Studies of predominantly Ashkenazi Jewish children with CHI suggest that the natural history of the disease is one of progressive glucose intolerance and clinical diabetes, possibly due to a slow progressive loss of β-cell function, and this may be due to the increased β-cell apoptosis, and, therefore, surgery may not be indicated in all patients [72]. Similarly, some patients with diazoxide-responsive CHI go on to develop diabetes mellitus in adulthood [73].
Near-total pancreatectomy is a major operation and is associated with a high incidence of diabetes mellitus later in life [74]. Clearly, surgery is indicated in those patients with severe diffuse disease who fail to respond to octreotide with frequent feeding regimens, and identification of this subgroup is important. The management of post-pancreatectomy diabetes mellitus is complicated by the fact that these children have pancreatic exocrine insufficiency, glucagon deficiency, and have residual unregulated insulin secretion, and some patients show resistance to hyperketonaemia and diabetic ketoacidosis [75]. Figure 5 is a suggested outline of the long-term medical and surgical management of patients with diazoxide-unresponsive CHI.
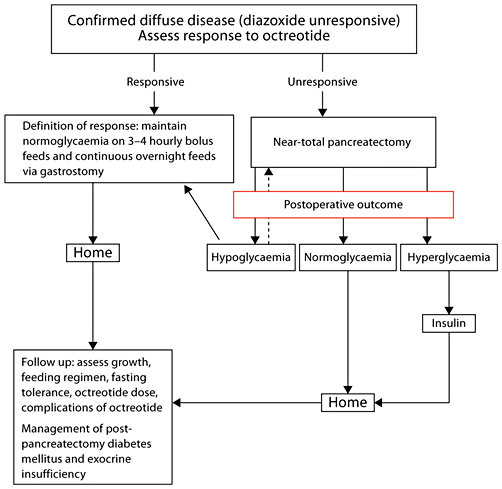
Outline of the long-term medical and surgical management of patients with diazoxide-unresponsive CHI. The broken arrow indicates that some patients may require a total pancreatectomy to control the most severe forms of CHI.
Outline of the long-term medical and surgical management of patients with diazoxide-unresponsive CHI. The broken arrow indicates that some patients may require a total pancreatectomy to control the most severe forms of CHI.
Conclusions
HH is a major cause of hypoglycaemia in the childhood period. Recognition and appropriate management of this type of hypoglycaemia are important to avoid long-term neurological consequences. The genetic mechanisms that lead to some forms of transient and CHI are beginning to be understood. Recent experience using 18F-L-dopa PET/CT scanning to distinguish diffuse from focal hyperinsulinism has completely changed the diagnostic and management approach to these patients. For the future, the management of medically unresponsive diffuse disease remains a challenge, and identifying the genetic mechanisms leading to both transient and persistent hyperinsulinism in the remaining 50% of the patients will provide novel insights into pancreatic β-cell physiology.
Acknowledgement
Research at the Institute of Child Health and Great Ormond Street Hospital for Children NHS Trust benefits from R & D funding received from the NHS Executive.
