

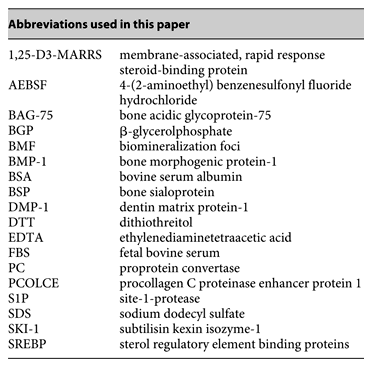
Abstract
The biochemical mechanism controlling nucleation of mineral crystals in developing bone, along with the growth and propagation of these crystals once formed, remains poorly understood. To define the nucleation mechanism, a proteomics analysis was begun on isolated biomineralization foci (BMF), sites of initial crystal nucleation in osteoblastic cell cultures and in primary bone. Comparative analyses of the protein profile for mineralized BMF with that for total osteoblast cultures revealed the latter were enriched in several proteins including BAG-75 and BSP, as well as fragments of each. When 12 protease inhibitors were added separately to UMR 106-01 osteoblastic cultures, only the serine protease inhibitor 4-(2-aminoethyl) benzenesulfonyl fluoride hydrochloride (AEBSF) blocked cleavage of BAG-75 and BSP, and prevented mineral crystal nucleation within BMF. Consideration of the specificities of the inhibitors tested and the fact that AEBSF inhibition was not dependent upon inclusion of FBS in the culture media indicated that mineral nucleation does not require serine protease plasmin, thrombin, kallikrein, urokinase, C1s or furin. In contrast, SKI-1 (S1P or site-1) is a membrane-bound serine protease inhibitable by AEBSF. We show here for the first time that mineralizing UMR 106 cells express a 98-kDa active, soluble form of SKI-1 within BMF. In contrast, nonmineralizing UMR cells appear to differentially process SKI-1 into smaller immunoreactive fragments (<35 kDa). These findings suggest that SKI-1 plays a direct or indirect role in assembly of functional nucleation complexes containing BAG-75 and BSP and their fragments, thus facilitating initial mineral nucleation within these biomineralization foci.
Introduction
Primary or woven bone is believed to be ‘disorganized’ by virtue of the appearance of its collagenous matrix. While controversy still exists over whether mineralization is initiated by collagen, by matrix vesicles, a combination of both or by other mechanisms, mineralization of primary bone is less well studied. Primary bone is an embryonic precursor of lamellar bone; however, fixed fractures heal and bone gained through biomechanical adaptation also form primarily via a woven bone intermediate [Turner et al., 1994; Mark et al., 2004]. Contrasted with compact lamellar bone, primary bone is distinguished by the speed of its formation and mineralization estimated to be 6 times faster [Kimmel and Jee, 1980]. A careful examination of noncollagenous components and cellular responses of primary and lamellar bone caused us to propose that each utilizes a different, but related, osteogenic program [Gorski, 1998]. We have identified a specific biomarker (bone acidic glycoprotein-75, BAG-75) in rapidly developing primary bone and osteoblastic cell cultures which spatially defines focal sites which will become mineralized up to 2 days later (biomineralization foci, BMF) [Gorski et al., 2004; Midura et al., 2004].
BMF are identifiable in several osteoblastic culture models where they appear as extracellular spherical 15- to 25-µm diameter complexes containing several size populations of vesicles [Midura et al., 2004]. In addition to BAG-75, bone sialoprotein (BSP) also becomes associated with BMF after addition of a phosphate source to the culture. BSP is a known nucleator of calcium phosphate crystal formation [Goldberg et al., 1996]. BAG-75 contains over 40 phosphate groups per mole, can bind up to 180 atoms of calcium per mole and is capable of self-associating into 12-nm fibrils stretching up to 1 µm in length [Chen et al., 1992]. A 50-kDa fragment of BAG-75 is also present in bone and serum [Gorski et al., 1990]. Interestingly, the serum level of the 50-kDa fragment appears to correlate with the rate of bone formation in vivo [Gorski, unpubl. result].
Most cells express subtilisin-like serine proteinases, the proprotein convertases (PCs), which mediate the proteolytic processing of secretory glycoproteins and the regulated release of membrane-bound transcription factors such as ATF6 and SREBPs [Seidah and Prat, 2002; Pasquato et al., 2006]. The prototypic basic amino acid-specific PC is furin [Seidah and Chretien, 1999], which was shown to activate BMP-1/Tolloid [Leighton and Kadler, 2003]. Activated BMP-1 has been shown to catalyze the cleavage of DMP-1 into 57- and 37-kDa fragments [Steiglitz et al., 2004].
Based on our initial studies [Gorski et al., 2004; Midura et al., 2004], we propose a unifying model for mineralization in primary bone that is initiated within BMF and regulated by osteoblastic cells (fig. 1). This model reconciles many of the existing ideas on mineralization into a single pathway that includes both active (cell-mediated) and passive (physicochemical) processes as well as matrix vesicle- and collagen-mediated mineralization mechanisms. Figure 1 summarizes this hypothesis, which provides a conceptual framework for our ongoing studies.

Proposed 2-stage model of biomineralization of primary bone. Immature BMF assemble from proteins and vesicles released from osteoblastic cells and represent the physical sites of initial mineralization nucleation within a developing or healing bone. This process is believed to be under cellular regulatory control. Growth and expansion of this initial mineral phase occurs when calcospherulite particles are subsequently released from mineralized BMF and seed the territorial collagenous matrix. This process is believed to be a passive physiochemical process. ECM = Extracellular matrix; AP = alkaline phosphatase; Pi = inorganic phosphate.
Proposed 2-stage model of biomineralization of primary bone. Immature BMF assemble from proteins and vesicles released from osteoblastic cells and represent the physical sites of initial mineralization nucleation within a developing or healing bone. This process is believed to be under cellular regulatory control. Growth and expansion of this initial mineral phase occurs when calcospherulite particles are subsequently released from mineralized BMF and seed the territorial collagenous matrix. This process is believed to be a passive physiochemical process. ECM = Extracellular matrix; AP = alkaline phosphatase; Pi = inorganic phosphate.
Experimental Procedures
Materials
Antibodies were from several sources: nonimmune rabbit IgG (EMD Biosciences, Inc.), anti-BAG-75 No. 503 (anti-peptide antibody) rabbit serum [Gorski et al., 1990], anti-SKI-1 antibody [Elagoz et al., 2002], anti-BAG-75 No. 504 (anti-protein antibody) rabbit serum [Gorski et al., 1990], anti-bone sialoprotein LF-100 antiserum (Larry Fisher, National Institute of Dental and Craniofacial Research, NIH) and monoclonal anti-BSP [WV1D1(9C5)] antibody (NIH Developmental Studies Hybridoma Bank, University of Iowa).
Cell Culture and Treatment with Protease Inhibitors
UMR 106-01 BSP cells were passaged and cultured at 37°C and 5% carbon dioxide as described previously [Wang et al., 2000] and updated briefly here. Cells were seeded at a density of 1.0 × 105 cells/cm2 in growth medium [Eagle’s MEM supplemented with Earle’s salts, 1% nonessential amino acids (Sigma-Aldrich Co.), 10 mM HEPES, pH 7.2, and 10% FBS (Hyclone)]. After 24 h, the medium was exchanged with growth medium containing 0.5% BSA (catalog No. A-1933; Sigma-Aldrich Co.) instead of FBS. Sixty-four hours after plating, the culture medium was exchanged with mineralization media [growth medium containing either 0.1% BSA or 10% FBS and 7 mM β-glycerolphosphate (BGP)]. Cultures were then incubated for an additional 24 h, at the end of which (88 h) the cells were either subjected to MTT assay or fixed in 70% ethanol and then extracted for protein. In some experiments, protease inhibitors, including the serine protease inhibitor (4-(2-aminoethyl)-benzenesulfonylfluoride hydrochloride (AEBSF; EMD Biosciences Inc.), were added to cultures at 64 h after plating in mineralization media. Alternatively, AEBSF was added at 44 h after plating; inhibitor was then removed and exchanged for mineralization media at 64 h and the amount of mineralization analyzed at 88 h.
Primary mouse osteoblasts were isolated from calvaria of 5- to 7-day-old mice using a modification of a published method [Huffman et al., 2007]. Calvaria were aseptically harvested and 4 sequential 20-min digests were performed in 0.05% trypsin/0.2% collagenase in Hanks’ balanced salt solution. Fractions 2 through 4 were pooled, centrifuged and resuspended in α-MEM containing 10% FBS, 2 mM L-glutamine, 100 µ/ml penicillin and 30 µg/ml gentamicin (α-growth medium). Per T-75-cm2 flask, 2 × 106 cells were plated and allowed to reach confluency (3–4 days). Confluent flasks were then trypsinized and plated into 12- or 24-well culture dishes for experiments at a density of 20,000 cells per cm2 growth area using media and supplements as described above. At confluency, the media were changed to α-MEM containing 5% FBS, 50 µg/ml ascorbic acid, 5 mM BGP and other supplements as described above. BGP was omitted from some wells which served as unmineralized controls. In order to test the effect of AEBSF, identical duplicate cultures were treated on days 3, 6 or 9 with 0.003–0.1 mM AEBSF. On day 12 after plating, one set of cultures was incubated with MTT as described below to determine cell viability. A second set of cultures was fixed on day 12 with 70% ethanol and processed for quantitative alizarin red S staining as described below.
MTT Assay
Culture wells were washed with Eagle’s MEM supplemented with Earle’s salts and then incubated with a solution of 0.5 mg/ml MTT (3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyl-2 H-tetrazolium bromide) in Eagle’s MEM for 1–2 h at 37°C [Huffman et al., 2007]. Residual MTT solution was removed, the cells disrupted by mixing briefly with dimethylsulfoxide, and free, reduced dye was read at 490 or 540 nm in a spectrophotometer.
Quantitation of Mineralization
After fixation in 70% ethanol, the cell layer was rinsed and stained with alizarin red S dye [Huffman et al., 2007]. The same procedure was also used for serum-depleted cultures with the following modified washing protocol, that is, the stained cell layer was rinsed once with 1 mM HEPES in nanopure water. A standard curve for alizarin red S dye was constructed for each analysis and the amount of bound dye per culture well was determined.
Statistical Methods
All statistical tests were performed using Sigma Stat 3.1 software (Systat Software Inc.). A one-way analysis of variance test was used to determine if a statistical difference existed between the viability of UMR 106-01 cultures or the amount of mineral deposited. Subsequent pair-wise multiple comparison tests were performed with the Student-Newman-Keuls or Kruskal-Wallis method.
Extraction of Cell Layer Fraction
Cells were dislodged by scraping and then extracted with 75-mM potassium phosphate buffer (pH 7.2), containing 10 mM CHAPS, 75 mM sodium chloride, 50 mM tetra sodium EDTA, 10 mM benzamidine hydrochloride, 2 mM DTT and 0.02% sodium azide for 1 h at 4°C. Each extract was then homogenized briefly using a motorized pestle and clarified by ultracentrifugation at 30,000 rpm for 1 h at 4°C in an SW 50.1 rotor prior to use. Conditioned media was immediately heated to 95°C for 5 min to inactivate protease activity and frozen at –80°C until analyzed.
Laser Capture Microscopy
UMR cells were grown as usual on Fisher Plus microscope slides (Fisher Scientific Inc.), fixed and stained with alizarin red S dye. Immediately prior to laser capture, slides were dehydrated through a graded series of ethanol washes and xylene rinses. Dried slides were stored at –20°C in a sealed box with desiccant until used. Mineralized BMF were collected onto standard caps using an Arturus Pixel IIe microscope. Collection films were pooled and stored in 70% ethanol at –20°C until approximately 6,200 BMF were collected. Laser microscopy-captured BMF were then mixed in 70% ethanol to dislodge the purple-stained particles which were then microfuged to remove the ethanol. BMF pellets were extracted twice sequentially over a 2-day period at 4°C with 1.1 ml of 0.1 M Tris-acetate buffer (pH 7.8) containing 0.5% octyl-glucoside, 0.05% SDS, 0.05 M EDTA and 0.02% sodium azide. Extracts were then dialyzed first against 0.01 M Tris-acetate buffer (pH 7.8) containing 8 M urea, 0.05% SDS, 0.1% octyl-glucoside, 0.05 M EDTA, and second against 0.01 M Tris-acetate buffer (pH 7.8) containing 8 M urea, 0.05% SDS and 0.1% octyl-glucoside. Controls consisted of glass slides containing the total cell layer fractions from +BGP or –BGP cultures; control slides were extracted using a similar protocol. The resultant dialyzed extracts were used for comparative blotting studies where identical protein amounts were loaded per gel lane.
Protein Determination
Protein concentration of BMF extracts was determined using the Non-Interfering Protein Assay by Geno Technology Inc.
Western Blotting and Chemiluminescence Detection
Extracts and media fractions prepared as described above were electrophoresed under reducing conditions on 4–20% linear gradient gels (ISC BioExpress) and electroblotted onto PVDF membranes (Millipore Corp.) for 2 h at 100 V [Huffman et al., 2007]. The transfer buffer was composed of 10 mM CAPS buffer (pH 11.0) containing 10% methanol. Blots were processed as described previously [Huffman et al., 2007]. Films were digitized using a flat bed scanner.
Results
Biomineralization Foci
Previous work has identified BMF as the sites of initial mineral crystal nucleation within osteoblastic cell cultures and in primary bone [Gorski et al., 2004; Midura et al., 2004; Huffmann et al., 2007]. Immunostaining and electron microscopy indicate that BMF are heterogeneous complexes of proteins as well as phospholipid-containing vesicles enriched in BAG-75 and BSP proteins. BMF are detectable in cultures prior to addition of phosphate; following addition of a phosphate source, nucleation occurs exclusively within BMF.
To identify the proteins regulating mineral nucleation, we have isolated mineralized BMF using laser capture microscopy and begun to characterize the BMF proteome [Huffmann et al., 2007]. Initial results have shown, as predicted, that the biomarkers BAG-75 and BSP are highly enriched within isolated BMF (fig. 2). This result validated the use of laser capture microscopy to purify mineralized BMF. Unexpectedly, BMF were also enriched in 50-kDa fragments of BAG-75 and BSP (fig. 2). Since these fragments were predominantly localized to BMF, this finding raised the possibility that proteolytic cleavage of these phosphoproteins is involved with mineralization.
![Fig. 2. Comparative immunostaining of laser capture microscope-isolated BMF versus total cell layer fractions demonstrates an enrichment of BAG-75 and BSP and their fragments. Protein extracts from laser microscope-captured BMF, cell layer fractions and a buffer control were electroblotted onto PVDF membrane and developed with anti-BAG-75 or anti-BSP antibodies. Arrows indicate 45- to 50-kDa fragments of BAG-75 and of BSP enriched in BMF extracts over that in the +CL total cell layer. The amount of protein loaded onto gel lanes was 6.5 µg (a–c) and 13 µg (d). d The blot was intentionally overdeveloped to detect the BSP fragment. Antibodies used: anti-BAG-75 protein antibody (No. 504), anti-BAG-75 peptide antibody (No. 503) and anti-BSP antibody (LF-100). BMF = Extract of laser microscope-captured BMF; +CL = extract of total cell layer from BGF-treated cultures; –CL = extract of total cell layer from cultures not treated with BGF; buffer = extraction buffer alone. From Huffman et al. [2007], reprinted with permission of the American Society for Biochemistry and Molecular Biology.](https://karger.silverchair-cdn.com/karger/content_public/journal/cto/189/1-4/10.1159_000151723/2/m_000151723_f02.jpeg?Expires=1716680124&Signature=GwSgXmrX59vj7Xwf16uT8KyXhQQjGkiCPuNyLn6zI-eedwZL3hv7F74eP5CdOOQ6slcw46rFTAcDKCOyv7E21PlLyoQvCsCh6RT4keYrvR2YqSG0vZ4ne9LmRSJKBaySi~48XOGfZtQKiFASXDqRaDFIpKsGxfRHWsrkS4U9bEbaNy18puWXKJty-N1aFIFzzii6QWb~oeZS~ux2F50yTGVU36yMMoY0pdyo-BsMzvbCSDRBZZ9Lv3r8zoU0GzdV3IbZJIKga6IzZ9VRXhpj0U-t1dK8gYjVB6rIq6v8dd9Mfaa4Hxv7WzgEbflM8bK2rCYKFWHtW8vznBXYijuOEQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Comparative immunostaining of laser capture microscope-isolated BMF versus total cell layer fractions demonstrates an enrichment of BAG-75 and BSP and their fragments. Protein extracts from laser microscope-captured BMF, cell layer fractions and a buffer control were electroblotted onto PVDF membrane and developed with anti-BAG-75 or anti-BSP antibodies. Arrows indicate 45- to 50-kDa fragments of BAG-75 and of BSP enriched in BMF extracts over that in the +CL total cell layer. The amount of protein loaded onto gel lanes was 6.5 µg (a–c) and 13 µg (d). d The blot was intentionally overdeveloped to detect the BSP fragment. Antibodies used: anti-BAG-75 protein antibody (No. 504), anti-BAG-75 peptide antibody (No. 503) and anti-BSP antibody (LF-100). BMF = Extract of laser microscope-captured BMF; +CL = extract of total cell layer from BGF-treated cultures; –CL = extract of total cell layer from cultures not treated with BGF; buffer = extraction buffer alone. From Huffman et al. [2007], reprinted with permission of the American Society for Biochemistry and Molecular Biology.
Comparative immunostaining of laser capture microscope-isolated BMF versus total cell layer fractions demonstrates an enrichment of BAG-75 and BSP and their fragments. Protein extracts from laser microscope-captured BMF, cell layer fractions and a buffer control were electroblotted onto PVDF membrane and developed with anti-BAG-75 or anti-BSP antibodies. Arrows indicate 45- to 50-kDa fragments of BAG-75 and of BSP enriched in BMF extracts over that in the +CL total cell layer. The amount of protein loaded onto gel lanes was 6.5 µg (a–c) and 13 µg (d). d The blot was intentionally overdeveloped to detect the BSP fragment. Antibodies used: anti-BAG-75 protein antibody (No. 504), anti-BAG-75 peptide antibody (No. 503) and anti-BSP antibody (LF-100). BMF = Extract of laser microscope-captured BMF; +CL = extract of total cell layer from BGF-treated cultures; –CL = extract of total cell layer from cultures not treated with BGF; buffer = extraction buffer alone. From Huffman et al. [2007], reprinted with permission of the American Society for Biochemistry and Molecular Biology.
Serine Protease Inhibitor AEBSF Specifically Inhibits Mineralization
In order to investigate the nature of the protease activity responsible and the relationship of BAG-75/BSP cleavage with mineral nucleation within BMF, we tested a series of 12 protease inhibitors (table 1) in the UMR 106-01 osteoblastic cell culture model. Specifically, individual inhibitors were added to confluent cultures at 64 h after plating and the amount of hydroxyapatite formed within BMF was scored 24 h later. UMR cultures are not competent to mineralize until 60 h after plating, reflecting an osteogenic differentiation process which leads to the production of spherical immature BMF structures enriched in BAG-75. Only one inhibitor, AEBSF, was found to interfere with mineral nucleation (table 1; fig. 3). AEBSF is a serine protease inhibitor which binds covalently, causing permanent inactivation. In view of the potential toxicity of AEBSF, we compared its inhibitory effect with that for overall cell viability (fig. 3a). While AEBSF specifically blocked nucleation of mineral crystals within BMF at 0.01–0.4 mM, cell viability was affected only above 1 mM. Alternatively, if cells were treated for 24 h with AEBSF, starting at 40 h after plating rather than at 64 h, the inhibitor was 10 times less effective in blocking subsequent mineralization scored at 88 h. Importantly, AEBSF also blocks mineralization of primary mouse calvarial osteoblastic cells with a similar sensitivity. Complete inhibition was observed whether AEBSF treatment was started on day 3, 6 or 9 after plating (data not shown), where mineralization occurs on day 12 (fig. 3b). Thus, mineralization mediated by osteoblastic cells in culture is associated with an AEBSF-inhibitable serine protease. Results from both primary and transformed osteoblastic cultures suggest a serine protease acts late in the process of mineral nucleation. Thus, mineral nucleation mediated by osteoblastic cells in culture is associated with an AEBSF-inhibitable serine protease.

![Fig. 3. AEBSF inhibits mineral nucleation both in UMR 106 osteoblastic cultures and in primary mouse calvarial osteoblasts. a With UMR cells, AEBSF blocks mineralization similarly in both serum-containing and serum-depleted conditions, while displaying higher effectiveness with mineralization-competent cultures. The amount of alizarin red S bound to calcium phosphate crystals within BMF and cell viability assessed with the MTT assay were plotted versus the concentration of AEBSF added to cultures. AEBSF was present twice during the culture model, that is, 44– 64 h and 64–88 h after plating. A 4-fold increase in sensitivity was observed in converting from serum-sufficient conditions to serum-depleted conditions, while a 10-fold increase in effectiveness was obtained when comparing 64–88 h versus 44–64 h cultures. For 64–88 h cultures: □ = MTT absorbance in serum-depleted conditions; █ = MTT absorbance in serum-containing media; ○ = amount of alizarin red bound in serum-depleted conditions; ● = amount of alizarin red bound in serum-containing media. For 44–64 h cultures: ▾ = amount of alizarin red bound in serum-containing media. MTT assay results for 44–64 h cultures were essentially identical to those for 64–88 h cultures and were omitted from the graph to maintain clarity. For the MTT cell viability assays, individual results (*) were significantly different from cultures treated with 1 mM AEBSF at a 99.9% confidence level. For alizarin red S assays on 64–88 h cultures, individual results were significantly different from cultures treated with 0.04 mM AEBSF (+) and with 0.01 mM AEBSF (#) at a 99.9% confidence level. For alizarin red S assays on 44–64 h cultures, individual results (**) were significantly different from cultures treated with 1 mM AEBSF at a 99.8% confidence level. Results shown are representative of 4 experiments. b AEBSF inhibits mineralization within nodules of primary mouse calvarial cultures without affecting viability. MTT assay results and the amount of alizarin red S bound to mineral deposits within cultures on day 12 are plotted versus the concentration of AEBSF added to cultures on day 9. □ = MTT absorbance in serum-containing media; ○ = amount of alizarin red bound in serum-containing media. For alizarin red S dye binding results for primary mouse calvarial cultures, untreated controls (‡) and 0.003 mM AEBSF wells (‡) were significantly different from those treated with higher concentrations of AEBSF at a 99.6% confidence level. For MTT assay results for primary mouse calvarial cultures, untreated controls (#), 0.003 mM AEBSF wells (#) and 0.03 mM AEBSF wells (#) were significantly different from those treated with 0.01 and 0.1 mM AEBSF at a 99.4% confidence level. Results depicted are representative of 3 experiments. Error bars refer to the standard deviation of the mean. UMR culture studies were carried out in triplicate, while primary culture studies were done in quadruplicate. All analyses are based on a one-way analysis of variance comparison using a Student-Newman-Keuls multiple comparison test. From Huffman et al. [2007], reprinted with permission of the American Society for Biochemistry and Molecular Biology.](https://karger.silverchair-cdn.com/karger/content_public/journal/cto/189/1-4/10.1159_000151723/2/m_000151723_f03a.gif?Expires=1716680124&Signature=xvOAMnBc9YU9XUtWymS15tYvxqh0w5aOU0B1u2AwUcfgxoo-CU3XKlzGZ7PGX8m8OUpuMQEQsUo5bSYs83CvaNT6DG2F3xOjPutlV2rQ2zslmZASsuP1Ik~tWFLgFWdi-V8biBkF6Sn6vCBruOZRsHomutUJGgs6nC2~ZCzJZkuzgdrr87IR41EsBpT8Da3b2QGOSOlicS-a2MaoC7mz~vIwOaFSox2vlDOE5jRzHciBO0d8PrWQ50GB9LoAkeAqf~-9tC-NUKPlQZ9VAFQsr11pvF4HKk8F5d23pfR33hGt-hlNc3Ec2OPr~5QXvSKtfO6nFL7QVy5KYVPB-5n8wg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
![Fig. 3. AEBSF inhibits mineral nucleation both in UMR 106 osteoblastic cultures and in primary mouse calvarial osteoblasts. a With UMR cells, AEBSF blocks mineralization similarly in both serum-containing and serum-depleted conditions, while displaying higher effectiveness with mineralization-competent cultures. The amount of alizarin red S bound to calcium phosphate crystals within BMF and cell viability assessed with the MTT assay were plotted versus the concentration of AEBSF added to cultures. AEBSF was present twice during the culture model, that is, 44– 64 h and 64–88 h after plating. A 4-fold increase in sensitivity was observed in converting from serum-sufficient conditions to serum-depleted conditions, while a 10-fold increase in effectiveness was obtained when comparing 64–88 h versus 44–64 h cultures. For 64–88 h cultures: □ = MTT absorbance in serum-depleted conditions; █ = MTT absorbance in serum-containing media; ○ = amount of alizarin red bound in serum-depleted conditions; ● = amount of alizarin red bound in serum-containing media. For 44–64 h cultures: ▾ = amount of alizarin red bound in serum-containing media. MTT assay results for 44–64 h cultures were essentially identical to those for 64–88 h cultures and were omitted from the graph to maintain clarity. For the MTT cell viability assays, individual results (*) were significantly different from cultures treated with 1 mM AEBSF at a 99.9% confidence level. For alizarin red S assays on 64–88 h cultures, individual results were significantly different from cultures treated with 0.04 mM AEBSF (+) and with 0.01 mM AEBSF (#) at a 99.9% confidence level. For alizarin red S assays on 44–64 h cultures, individual results (**) were significantly different from cultures treated with 1 mM AEBSF at a 99.8% confidence level. Results shown are representative of 4 experiments. b AEBSF inhibits mineralization within nodules of primary mouse calvarial cultures without affecting viability. MTT assay results and the amount of alizarin red S bound to mineral deposits within cultures on day 12 are plotted versus the concentration of AEBSF added to cultures on day 9. □ = MTT absorbance in serum-containing media; ○ = amount of alizarin red bound in serum-containing media. For alizarin red S dye binding results for primary mouse calvarial cultures, untreated controls (‡) and 0.003 mM AEBSF wells (‡) were significantly different from those treated with higher concentrations of AEBSF at a 99.6% confidence level. For MTT assay results for primary mouse calvarial cultures, untreated controls (#), 0.003 mM AEBSF wells (#) and 0.03 mM AEBSF wells (#) were significantly different from those treated with 0.01 and 0.1 mM AEBSF at a 99.4% confidence level. Results depicted are representative of 3 experiments. Error bars refer to the standard deviation of the mean. UMR culture studies were carried out in triplicate, while primary culture studies were done in quadruplicate. All analyses are based on a one-way analysis of variance comparison using a Student-Newman-Keuls multiple comparison test. From Huffman et al. [2007], reprinted with permission of the American Society for Biochemistry and Molecular Biology.](https://karger.silverchair-cdn.com/karger/content_public/journal/cto/189/1-4/10.1159_000151723/2/m_000151723_f03b.gif?Expires=1716680124&Signature=oYo8~qkJsgaI0bidUbkxm1uTN-b2ZMRcFi4B8S5UYzVVt~4QFRwXNZSD72daN3c7Ox4Zw2Fe6mDAIICURGMyq-b5YXC9zaD1o3H0vfzXbx94Qt8eQyMblm3Z216taKjNhq8MU59PUmGBAxdLOJxJLYvsNL5my1aNAjj-PO5SljC5WcJqhPlCKXd7mXOFfLPBdRYxARSlgqvobcwj3KRkT0TbzZd3ljhM95SjWX1gdrIz1Y0hfMhmoZj1ePNFTIGL2lo1fWneBQHElOyYh139WjjixtOS0DOWVpn5ShhJFdqtGCPBCU1yng4Yz89amApTqwpWsE7WwZ~leNCCTAJObQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
AEBSF inhibits mineral nucleation both in UMR 106 osteoblastic cultures and in primary mouse calvarial osteoblasts. a With UMR cells, AEBSF blocks mineralization similarly in both serum-containing and serum-depleted conditions, while displaying higher effectiveness with mineralization-competent cultures. The amount of alizarin red S bound to calcium phosphate crystals within BMF and cell viability assessed with the MTT assay were plotted versus the concentration of AEBSF added to cultures. AEBSF was present twice during the culture model, that is, 44– 64 h and 64–88 h after plating. A 4-fold increase in sensitivity was observed in converting from serum-sufficient conditions to serum-depleted conditions, while a 10-fold increase in effectiveness was obtained when comparing 64–88 h versus 44–64 h cultures. For 64–88 h cultures: □ = MTT absorbance in serum-depleted conditions; █ = MTT absorbance in serum-containing media; ○ = amount of alizarin red bound in serum-depleted conditions; ● = amount of alizarin red bound in serum-containing media. For 44–64 h cultures: ▾ = amount of alizarin red bound in serum-containing media. MTT assay results for 44–64 h cultures were essentially identical to those for 64–88 h cultures and were omitted from the graph to maintain clarity. For the MTT cell viability assays, individual results (*) were significantly different from cultures treated with 1 mM AEBSF at a 99.9% confidence level. For alizarin red S assays on 64–88 h cultures, individual results were significantly different from cultures treated with 0.04 mM AEBSF (+) and with 0.01 mM AEBSF (#) at a 99.9% confidence level. For alizarin red S assays on 44–64 h cultures, individual results (**) were significantly different from cultures treated with 1 mM AEBSF at a 99.8% confidence level. Results shown are representative of 4 experiments. b AEBSF inhibits mineralization within nodules of primary mouse calvarial cultures without affecting viability. MTT assay results and the amount of alizarin red S bound to mineral deposits within cultures on day 12 are plotted versus the concentration of AEBSF added to cultures on day 9. □ = MTT absorbance in serum-containing media; ○ = amount of alizarin red bound in serum-containing media. For alizarin red S dye binding results for primary mouse calvarial cultures, untreated controls (‡) and 0.003 mM AEBSF wells (‡) were significantly different from those treated with higher concentrations of AEBSF at a 99.6% confidence level. For MTT assay results for primary mouse calvarial cultures, untreated controls (#), 0.003 mM AEBSF wells (#) and 0.03 mM AEBSF wells (#) were significantly different from those treated with 0.01 and 0.1 mM AEBSF at a 99.4% confidence level. Results depicted are representative of 3 experiments. Error bars refer to the standard deviation of the mean. UMR culture studies were carried out in triplicate, while primary culture studies were done in quadruplicate. All analyses are based on a one-way analysis of variance comparison using a Student-Newman-Keuls multiple comparison test. From Huffman et al. [2007], reprinted with permission of the American Society for Biochemistry and Molecular Biology.
AEBSF inhibits mineral nucleation both in UMR 106 osteoblastic cultures and in primary mouse calvarial osteoblasts. a With UMR cells, AEBSF blocks mineralization similarly in both serum-containing and serum-depleted conditions, while displaying higher effectiveness with mineralization-competent cultures. The amount of alizarin red S bound to calcium phosphate crystals within BMF and cell viability assessed with the MTT assay were plotted versus the concentration of AEBSF added to cultures. AEBSF was present twice during the culture model, that is, 44– 64 h and 64–88 h after plating. A 4-fold increase in sensitivity was observed in converting from serum-sufficient conditions to serum-depleted conditions, while a 10-fold increase in effectiveness was obtained when comparing 64–88 h versus 44–64 h cultures. For 64–88 h cultures: □ = MTT absorbance in serum-depleted conditions; █ = MTT absorbance in serum-containing media; ○ = amount of alizarin red bound in serum-depleted conditions; ● = amount of alizarin red bound in serum-containing media. For 44–64 h cultures: ▾ = amount of alizarin red bound in serum-containing media. MTT assay results for 44–64 h cultures were essentially identical to those for 64–88 h cultures and were omitted from the graph to maintain clarity. For the MTT cell viability assays, individual results (*) were significantly different from cultures treated with 1 mM AEBSF at a 99.9% confidence level. For alizarin red S assays on 64–88 h cultures, individual results were significantly different from cultures treated with 0.04 mM AEBSF (+) and with 0.01 mM AEBSF (#) at a 99.9% confidence level. For alizarin red S assays on 44–64 h cultures, individual results (**) were significantly different from cultures treated with 1 mM AEBSF at a 99.8% confidence level. Results shown are representative of 4 experiments. b AEBSF inhibits mineralization within nodules of primary mouse calvarial cultures without affecting viability. MTT assay results and the amount of alizarin red S bound to mineral deposits within cultures on day 12 are plotted versus the concentration of AEBSF added to cultures on day 9. □ = MTT absorbance in serum-containing media; ○ = amount of alizarin red bound in serum-containing media. For alizarin red S dye binding results for primary mouse calvarial cultures, untreated controls (‡) and 0.003 mM AEBSF wells (‡) were significantly different from those treated with higher concentrations of AEBSF at a 99.6% confidence level. For MTT assay results for primary mouse calvarial cultures, untreated controls (#), 0.003 mM AEBSF wells (#) and 0.03 mM AEBSF wells (#) were significantly different from those treated with 0.01 and 0.1 mM AEBSF at a 99.4% confidence level. Results depicted are representative of 3 experiments. Error bars refer to the standard deviation of the mean. UMR culture studies were carried out in triplicate, while primary culture studies were done in quadruplicate. All analyses are based on a one-way analysis of variance comparison using a Student-Newman-Keuls multiple comparison test. From Huffman et al. [2007], reprinted with permission of the American Society for Biochemistry and Molecular Biology.
SKI-1 Serine Protease Is Present in BMF
Figure 4 depicts the results of Western blotting with anti-SKI-1 N-terminal antibody and UMR 106-01 cell culture fractions. Specifically, total cell extracts from phosphate-supplemented and control cultures were compared with that for the laser capture microscope-purified BMF. It is apparent that SKI-1 is present in all mineralizing samples as a 98-kDa soluble enzyme (fig. 4, +CL and BMF); the predominant forms in nonmineralizing cultures are smaller than 35 kDa (fig. 4, –CL). Under normal conditions, SKI-1 resides in the cis/medial Golgi as an approximately 106-kDa active transmembrane form [Seidah and Chretien, 1999; Elagoz et al., 2002]. In specific circumstances, SKI-1 is transported to the plasma membrane, autocatalytically shed as an approximately 98-kDa catalytically active, soluble enzyme [Elagoz et al., 2002; Pullikotil et al., 2007]. The amount of 98-kDa form detected is similar in the total culture extracts and in the purified BMF preparation. While not indicative of a quantitative enrichment of SKI-1 to BMF complexes, our results demonstrate its association with structures mediating initial mineral nucleation.

Immunoblotting demonstrates the presence of SKI-1 in BMF and UMR 106 cultures. UMR cells were cultured on glass slides and resultant BMF were stained with alizarin red S dye and purified using laser capture microscopy (see Experimental Procedures). As controls, the total cell layer fraction from a mineralized culture (+CL) and an unmineralized culture (–CL) were extracted separately and subjected to SDS-PAGE and immunoblotting along with laser microscopy-captured BMF or buffer alone. Identical amounts of protein were applied to each lane and probed with anti-SKI-1 antibodies.
Immunoblotting demonstrates the presence of SKI-1 in BMF and UMR 106 cultures. UMR cells were cultured on glass slides and resultant BMF were stained with alizarin red S dye and purified using laser capture microscopy (see Experimental Procedures). As controls, the total cell layer fraction from a mineralized culture (+CL) and an unmineralized culture (–CL) were extracted separately and subjected to SDS-PAGE and immunoblotting along with laser microscopy-captured BMF or buffer alone. Identical amounts of protein were applied to each lane and probed with anti-SKI-1 antibodies.
Discussion
Serine proteases play a critical role in the activation and regulation of a number of biological processes in the extracellular environment, for instance, blood coagulation and complement activation. In this context, we hypothesize that the observed fragmentation of BAG-75 and BSP serves to activate their functional role(s) within BMF. While BSP is known to nucleate hydroxyapatite crystals, previous work has not shown a requirement for proteolytic activation [Goldberg et al., 1996]. We speculate that final assembly of nucleating complexes within BMF may require proteolytic fragmentation of BAG-75 and BSP. In all, we have identified 5 proteins whose cleavage is inhibited by AEBSF [Huffman et al., 2007].
PCs are comprised of 3 different groups of enzymes in terms of their substrate specificities [Seidah et al., 2006]. The 7 basic amino acid-specific convertases (PC1/3, PC2, furin, PC4, PC5/6, PACE4 and PC7), of which furin is the prototype, cleave preferentially following single or dibasic sequences following the motif (R/K)-Xn-(R/K)↓, where n = 0, 2, 4 or 6 amino acids, the 2 other convertases SKI-1/S1P and PCSK9 cleave at nonbasic residues after the motifs R-X-hydrophobic-X↓ [Pasquato et al., 2006] and VFAQ↓ [Benjannet et al., 2004; Seidah et al., 2006], respectively. Analysis of BSP, membrane-associated, rapid response steroid-binding (1,25-D3-MARRS) receptor and procollagen C-proteinase enhancer protein 1 (PCOLCE), 3 proteins whose cleavage is blocked by AEBSF [Huffman et al., 2007], reveals each contains several SKI-1 candidate cleavage sequences which appear capable of generating the observed fragments [Huffman, Seidah and Gorski, unpubl. results]. SKI-1 is a type I membrane-bound protease synthesized as a 128-kDa zymogen that is autocatalytically activated into transmembrane 106-kDa forms and secreted soluble forms of approximately 98 kDa [Seidah et al., 1999; Elagoz et al., 2002], both of which are inhibitable by AEBSF [Okada et al., 2003; Basak et al., 2004].
Evidence consistent with SKI-1’s identity as the AEBSF-susceptible protease involves its known sensitivity to AEBSF, its association with mineralized BMF and its expression by mineralizing UMR cells as a predominant functionally active soluble enzyme of approximately 98 kDa missing its C-terminal membrane-spanning domain. In UMR cells not treated with BGP, the relative content of active SKI-1 of approximately 98 kDa is diminished and, instead, the prominent immunoreactive bands are <35 kDa (fig. 4). This suggests that SKI-1 itself can be proteolytically processed, particularly under nonmineralizing conditions, as was also observed in embryonic kidney cells [Pullikotil et al., 2007]. In contrast, results with specific inhibitors indicate that functional plasmin, thrombin, kallikrein, C1s complement protease and urokinase plasminogen activator are not required for mineral crystal nucleation within BMF (table 1).
In conclusion, our studies implicate SKI-1 as a serine protease-regulating mineral crystal nucleation within BMF complexes. Future studies will use siRNA knockdown and SKI-1 plasmid transfection approaches to conclusively identify SKI-1 as the AEBSF-susceptible protease associated with mineral nucleation in primary bone.
Acknowledgements
Supported by grant No. R01-AR052775 from the NIH NIAMS (to J.P.G.), a CIHR grant No. MOP 36496 and a Canada Research Chair No. 201652 (to N.G.S.).
