

Abstract
Individuals within a species have genetic differences which ultimately result in the spectrum of phenotypic variation that we observe. Genetic variation exists at the nucleotide level in the form of single nucleotide polymorphisms (SNPs), and at a structural level as inversions, deletions and amplifications of larger stretches of nucleotides. Profiling of human and mouse genomes has identified numerous genomic segmental copy number variations (CNVs) throughout these genomes. Since inbred mice are widely used laboratory models for the study of both normal and disease biology, it is crucial that we understand the full scope of genetic variation, including CNVs, within these animals. These genetic differences can inform us about the history of a population or species, enlighten us on gene function, and guide our selection of a model system for the study of human disease.
The mouse as a model organism
The use of mice as research animals dates back to at least the 1800s. Their utility increased greatly at the turn of the 20th century with the development of the first characterized mouse strains by William Castle. This was soon followed by the creation of the first inbred mouse strain by Castle’s student Clarence Little in 1909 (Paigen, 2003; Eisen, 2005). Contemporaneously, one of the first uses of mice to study human disease was the crossing of ‘fancy mice’ with ‘common’ laboratory mice to understand the heritability of resistance to transplanted tumor growth by E.E. Tyzzer (Paigen, 2003). Tyzzer’s experiments were greatly complicated, however, by the genetic heterogeneity of his subjects. Using his new inbred mouse strain, Little solved this problem and conclusively demonstrated the Mendelian inheritance of transplanted tumor resistance loci.
This first strain, named DBA for the presence of the d (dilute), b (brown), and a (agouti) coat color alleles, was produced in the same manner as other inbred mouse strains that would come after, through repeated brother-sister crosses (and sometimes offspring-parent crosses) for at least 20 generations (Beck et al., 2000). This laboratory manipulation created a unique organism, a diploid mammal which simulated monoploidy by being homozygous at every (or nearly every) allele in the genome. These two characteristics, the phenotypic complexity of a mammal and a relatively static homozygous genetic background, made inbred mouse strains uniquely powerful models in which genetic studies of complex phenotypes could be carried out. In the years since the DBA strain was first generated, over 450 other mouse lines have been bred, although many of the most commonly used strains trace their history back almost as far as DBA (Beck et al., 2000).
Researchers have found proxies for various human diseases among this large number of disparate mouse strains, each with its own unique assortment of alleles and the phenotypes to which they give rise. While first used for studying cancer and immunology, the use of inbred mouse strains has become common in most, if not all, disease fields including the fields of study of developmental disorders, metabolic diseases, and neurologic and behavioral disorders (Bedell et al., 1997). In metabolic disease research, for example, strains have been identified with varying degrees of resistance or sensitivity to spontaneous and diet-induced diabetes, obesity, and atherosclerosis (Clee and Attie, 2007). These mice have long been used for Quantitative Trait Locus (QTL) mapping through the crossing of susceptible and resistant strains followed by serial back-crossing to identify the responsible genetic loci. More recently, rather than having to perform laborious back-crosses, the in silico technique of Whole Genome Analysis (WGA) has been developed (Grupe et al., 2001; Smith et al., 2003; Liao et al., 2004; Pletcher et al., 2004; Wang et al., 2005). In WGA, known sequence polymorphisms in existing inbred mouse strains are the foundation for the analysis. This technique opens the door to a much more time-efficient way to identify gene variants that affect phenotype, but it depends on prior characterization of a very large number of sequence polymorphisms. Fortunately, considerable work has already gone into cataloging large numbers of single-nucleotide polymorphisms (SNPs; Sherry et al., 1999) and this data served as the foundation for the studies referenced.
The state of CNV research in the mouse
Despite the focus on SNPs for genetic mapping, they are not the only type of sequence polymorphism known. Although less common than SNPs, there exist amplifications and deletions of segments of the genome ranging from 1 kb to many megabases, known as segmental copy number variations (CNVs). CNVs may play an important role in both the range of normal mammalian phenotypic differences (Freeman et al., 2006) as well as in the etiology of disease (Gonzalez et al., 2005; Aitman et al., 2006). The potentially dramatic effects of large scale CNVs on human health have long been understood based on the identification of gross karyotype alterations (Emanuel and Shaikh, 2001; Shaw and Lupski, 2004), but it is only recently that a fuller picture of CNV content in mammalian genomes has started to emerge. With the advent of modern genomic technology, particularly array-based Comparative Genomic Hybridization (aCGH), it has become possible to do large scale surveys of the CNV content of genomes (Pinkel et al., 1998; Barrett et al., 2004). A number of researchers have done this work in humans (Sebat et al., 2004; Sharp et al., 2005; Redon et al., 2006; Zogopoulos et al., 2007; Jakobsson et al., 2008), identifying many hundreds of CNVs across diverse sets of individuals. Likewise, aCGH analysis has been brought to bear on the murine genome, exploring the CNV content of various sets of inbred mouse strains (Li et al., 2004; Snijders et al., 2005; Cutler et al., 2007; Egan et al., 2007; Graubert et al., 2007; She et al., 2008; Watkins-Chow and Pavan, 2008).
The earlier work on identifying mouse CNVs performed by Li et al. (2004; hereafter referred to as Li) and Snijders et al. (2005; hereafter Snijders) used BAC-based CGH arrays containing 19,200 BAC probes and 2,069 BAC probes, respectively. Li identified 347 BACs that probed genomic regions with apparent copy-number variations in 14 inbred mouse strains, using C57BL/6 as the reference strain. Snijders identified 79 BACs that probed apparent CNVs in seven inbred and one outbred mouse strain, using FVB as the reference strain. Unfortunately, due to the use of BAC-based arrays with low to medium genomic resolution, from 0.16 Mb per probe to 1.5 Mb per probe for these two studies, the actual borders of the CNVs were ill-defined as was the basic fact of whether or not adjacent altered probes represented one contiguous CNV. More worrisome, however, was the observation that comparing the results of these two studies revealed ‘no obvious overlap’ between the CNV-defining BAC probes (Snijders et al., 2005).
The most recent large-scale mouse CNV surveys moved away from using BAC-based aCGH to oligo-based aCGH (Cutler et al., 2007; Graubert et al., 2007; She et al., 2008; hereafter referred to as Cutler, Graubert, and She, respectively). Graubert compared 20 mouse strains to C57BL/6 using high-density NimbleGen oligonucleotide arrays. These arrays had over 388,000 probes with a density of more than one probe per 8 kb. Cutler, which was work done by our group, used high-density Agilent oligonucleotide arrays with approximately 244,000 probes to survey 41 strains of inbred mouse compared to C57BL/6 – the most comprehensive analysis done to date. She used a sequencing method along with an aCGH analysis of 15 strains on NimbleGen arrays. There was broad agreement between the Graubert and Cutler oligo-aCGH-based studies, with 67% of 72 CNV loci identified by Graubert also identified by Cutler (Cutler et al., 2007). However, neither of these two studies identified much, if any, overlap with the two BAC-aCGH-based analysis. For example, the Graubert CNVs matched only 7% (17 out of 238) and 4% (3 out of 74) of the CNV loci from Li and Snijders, respectively (Graubert et al., 2007). The lack of concordance between either of the BAC-based analyses and any of the other studies probably derives from a combination of factors: the lower density of probes on those arrays makes it more likely that they will completely miss a large number of CNVs; BAC-identified CNVs are often identified from altered hybridization intensity of just a single probe; the greater length of the individual probes increases the chance that results will be confounded by cross-hybridization to multiple genomic regions; BACs are subject to recombination, misidentification, and mismapping to the genome at any of the many steps between the creation of the BAC library in bacteria to the spotting of the BAC probes onto arrays. In addition, we have observed that ‘home made’ non-commercial microarrays rarely, if ever, approach the quality and reproducibility found in commercially produced arrays (data not shown). Regardless of the ultimate source of the lack of reproducibility of the BAC-aCGH-based CNV results, it is clear that those initial findings must be treated with caution.
Although most of the CNVs identified by Graubert are also identified by Cutler, the reverse is not true. Only 26% of the 2,094 individual CNVs mapped by Cutler fall within CNV loci as defined in Graubert (Cutler et al., 2007). We believe that this is due to a more conservative CNV-calling algorithm used by Graubert coupled with a compressed range of fold-change amplitudes that we have previously observed with the NimbleGen platform (data not shown and see below). It remains to be seen how the concordance would change if a more sensitive algorithm is used to analyze the data from Graubert.
The CNV landscape in the mouse
Since Cutler is the most comprehensive analysis of mouse CNVs to date in terms of number of strains analyzed and has revealed the greatest number of CNVs, that body of data will be used for further discussion here, except where otherwise noted. That study identified 2,096 CNVs across 41 inbred mouse strains. This set of CNVs can be simplified by collapsing all overlapping CNVs between all strains into a set of 591 CNV loci, of which 224 loci contain CNVs from multiple mouse strains. The distribution of the remaining 367 singleton CNVs in terms of both length and amplitude is broadly similar to the 224 overlapping CNVs, though showing a statistically significant excess of lengths shorter than 10 kb (data not shown). Examining the full set of CNVs shows a mean length of 197 kb, close to the 323 kb mean length of CNVs identified in Graubert once those CNVs are mapped to the NCBI Build 36 of the mouse genome. The maximum size of detected CNVs on the autosomal chromosomes is in the 3 to 4 Mb range, although one CNV was found to extend over 8.6 Mb across the XMR locus on the X chromosome of Spret/EiJ. While there is no natural minimum limit to the length of CNVs, the density of probes on the microarrays used and the requirement of generally three or more adjacent perturbed probes imposes a technical minimum limit of approximately 36 bp. The smallest CNV actually detected is 62 bp long, although less than 0.3% of CNVs found were under 1 kb in length. This distribution of CNV lengths leads to an average of 10.1 Mb, or 0.38%, of the mouse genome being found within CNVs, with a maximum CNV content of 36.2 Mb, or 1.38%, found in Spret/EiJ.
The detected mouse CNVs are expected to have fold-change amplitudes that correspond to whole-number ratios. For example, a segmental duplication should have a fold change of 2 (a ratio of 2:1) and a segmental triplication should have a fold change of 3 (3:1), while a segmental deletion of a non-repeated sequence should have a negative-infinite fold change (0:1). This data should therefore be slightly easier to interpret than human CNV data where hemizygosity is an issue, and much easier to interpret than tumor CNV data where the DNA is isolated from samples that are generally contaminated with normal tissue resulting in a mixture of analyzed genomes. In fact, a histogram of the amplitudes of the identified CNVs matches closely what one would expect based on ratios of 1 to 3 diploid copies of genome segments, with the most prominent peaks corresponding to ratios of 0:1 (or –20-fold in this data set where extremely large and small fold-change values were truncated at 20-fold and –20-fold, respectively), 2:1, and 1:2 (Fig. 1). A similar analysis of the CNVs from Graubert shows a greatly compressed range of fold-change values, similar to what we have personally noted when working with NimbleGen arrays (data not shown), with fold-change amplitudes ranging only from –3.2-fold to 2.4-fold.

A comparison of amplitude histograms of CNVs from Cutler and Graubert. The smoothed histograms of the log2 fold-change values of the CNV datasets from Cutler (all significant CNVs; thick line) and Graubert (all CNVs with an absolute log2 amplitude >0.5; dashed line) are shown. The positions of whole-number ratio changes in copy number are indicated in parenthesis on the X-axis. Deletions are truncated at a minimum fold-change of 1/20 (log2 = –4.3). The two curves are scaled for plotting and the peak at –4.3 is off scale.
A comparison of amplitude histograms of CNVs from Cutler and Graubert. The smoothed histograms of the log2 fold-change values of the CNV datasets from Cutler (all significant CNVs; thick line) and Graubert (all CNVs with an absolute log2 amplitude >0.5; dashed line) are shown. The positions of whole-number ratio changes in copy number are indicated in parenthesis on the X-axis. Deletions are truncated at a minimum fold-change of 1/20 (log2 = –4.3). The two curves are scaled for plotting and the peak at –4.3 is off scale.
The amplitude histogram also reveals peaks that correspond to CNV amplitudes best explained as the whole number ratios 3:2, 2:3, 1:2, and 1:3. An example of a 1:2 ‘partial amplitude’ deletion – as opposed to a ‘full’ deletion where there are no remaining copies of the locus – is found on chromosome 14 between positions 68206900 and 68418187 (genomic coordinates based on the Feb. 2006 version of the NCBI mouse genome assembly; Fig. 2). Plotting the fold-change values across this region shows a deletion with a mean fold-change of –2.2 across all strains except for C57BL/6J. As expected, probes for C57BL/6J are unchanged when compared to the reference data – other C57BL/6J samples – with a mean fold-change of 1.0. The most parsimonious explanation for this phenomenon is the presence of two diploid copies of this genomic region in C57BL/6J and only one copy in all the other strains tested. However, a sequence search using a sampling of 29 of the microarray probes in this region reveals only one genomic match per probe in the C57BL/6-based mouse genome sequence (data not shown). Therefore, this and the other ‘partial amplitude’ deletions likely reveal regions in the published mouse genome sequence where duplicated loci – including gene-rich, euchromatic regions such as the chromosome 14 locus shown here – have been misassembled as unique loci. The sequencing-based analysis in She supports this conclusion by also identifying a large amount of duplicated sequence in the C57BL/6J genome (She et al., 2008). Likely, both aCGH data and more sophisticated sequence analysis will be important information sources to help guide and correct sequence assemblies.

A ‘partial amplitude’ deletion on murine chromosome 14. The log2 fold-change aCGH data for 42 inbred mouse strains compared to C57BL/6J is plotted for a region on chromosome 14 from nucleotide position 68206900 to 68418187. Data for C57BL/6J is shown in black while all other strains are shown in color. The locations of expected fold-changes based on whole-number copy losses are shown with dotted lines. The positions of known genes in this locus are shown at the top.
A ‘partial amplitude’ deletion on murine chromosome 14. The log2 fold-change aCGH data for 42 inbred mouse strains compared to C57BL/6J is plotted for a region on chromosome 14 from nucleotide position 68206900 to 68418187. Data for C57BL/6J is shown in black while all other strains are shown in color. The locations of expected fold-changes based on whole-number copy losses are shown with dotted lines. The positions of known genes in this locus are shown at the top.
Sequence content of mouse CNVs
It is important to understand the nature of that portion of the genome which is found in CNVs, both to understand the processes that give rise to CNVs as well as their potential impacts on biology. Short tandem repeats that make up microsatellite sequences as well as repeated sequences on the order of several 10s of kbs in length have been observed to expand or contract in number through processes such as non-reciprocal recombination and gene conversion (Richard and Pâques, 2000; Read et al., 2004). Graubert explored whether segmental duplications, repeated regions of at least 90% identity, were associated with mouse CNVs and did, in fact, find such a correlation with 47.5% of 80 CNVs overlapping known regions of segmental duplication. Since our analysis of CNV amplitudes suggests that there exist previously unknown regions of high identity repeats, a true characterization of the association between CNVs and tandem segmental repeats is yet to be determined. An analysis of the association between repeats, including both simple (e.g. microsatellites) and complex (e.g. LINES, SINES) repeats, and the larger CNV set from Cutler, in contrast, shows significant enrichment of low-repeat-content DNA in deletions, 54% greater than would be expected if there were no association, coupled with a highly significant enrichment of high-repeat-content DNA outside of CNVs (Cutler et al., 2007). The reasons for this are unclear and warrant further analysis.
A potential source of false-positive deletion CNV calls from aCGH data may be the presence of SNPs in the sequences probed by the microarray oligos. However, the overall distribution of CNV amplitudes (Fig. 1) argues against this hypothesis, as does the lack of correlation between SNP content and CNV content in mouse strains (Cutler et al., 2007). This can also be tested by comparing the genomic locations of SNPs with the locations of CNVs. Interestingly, genomic regions of low SNP content (less than or equal to 0.5 known SNPs per kb) were very strongly enriched in both deletion CNVs (305% of the amount expected with no association) and amplification CNVs (239% of expected). As with the association with low-repeat content, the reasons for this SNP association are unclear and may be due to both technical and biological causes.
Perhaps of greatest interest is the association between genes and CNVs. Both intergenic regions and CpG-island-poor genomic regions are enriched in deletion CNVs with strong statistical significance (136% and 105% of expected, respectively) while genes and CpG islands are both enriched outside of CNVs. Amplification CNVs show enrichment of neither genes nor intergenic sequence and a modest enrichment of CpG-island-poor regions, while pseudogenes are strongly enriched in deletion CNVs. These data together suggest that the rate of formation of deletion CNVs is decreased in gene-rich regions and/or mice experience decreased viability or fitness when such events occur, unlike the situation with gene amplifications or pseudogene deletions which would generally be expected to be phenotypically neutral. Additionally, genes associated with known heritable diseases in humans can be found in amplification CNVs, but rarely in deletion CNVs (discussed in more detail below). Genes with up to two known paralogs in mice are significantly enriched (103%) outside of CNVs while genes with many paralogs (>5), and therefore with a biology or genomic structure that increases their propensity to have altered copy numbers, are indeed strongly enhanced in both deletion CNVs (353%) and amplification CNVs (230%).
The gene annotation provided by the Gene Ontology (GO) consortium (Ashburner et al., 2000) allows us to look for enrichment or exclusion of gene functions in CNVs. Both Cutler and Graubert find broadly similar results, with pheromone receptor and olfactory receptor-related functions, respectively, being strongly enriched in CNVs. These genes are found in large families marked by rapid evolution, as evidenced by the presence of many pseudogenes and a highly variable gene content between species, and are likely under reduced evolutionary pressure. Another gene function which is identified as CNV-enriched in both studies is defense response, including genes such as chemokine receptors, histocompatibility receptors, interferons, defensins, and killer cell-like receptors. As with pheromone and olfactory receptor genes, many of these defense response genes are found in large, variable families which, in the aseptic laboratory environment, are likely under reduced selective pressure. Cutler also identifies the related antigen binding, processing, and presentation functions enriched in both amplifications and deletions. These particular results must be viewed with caution, however, since they include genes involved in somatic-level rearrangements in lymphoid cells and thus may not represent germ-line CNVs. In contrast to the types of genes enriched in CNVs, many essential gene functions are found enriched outside of CNVs, including transcription, cell cycle, and protein folding functions.
CNVs and mouse evolution
We have already noted the inverse relationship between the distribution of CNVs and SNPs. One possible contributor to this phenomenon is a different phylogenic history of SNP-containing vs. CNV-containing genomic regions. For example, if two types of genetic changes arise with differing frequency, the one which arises less frequently may probe more deeply into history while the one which arises more frequently may be a shorter-term marker of a strain’s lineage. Additionally, the randomness inherent in the chromosomal contributions of an organism’s two parents coupled with the smaller numbers of CNVs as compared to SNPs makes it more likely that CNV content will fluctuate more across generations. To test whether there is a difference in the phylogenic history of SNPS and CNVs, we used the SplitsTree program (Huson, 1998) to generate a ‘hybridization tree’ based on both SNP and CNV data (Fig. 3). The SNP-based tree appears to be a good representation of the known history of these strains (Beck et al., 2000). The tree based on CNV data is broadly similar to the SNP tree, as can be seen by noting the colors of the major sections of the SNP tree carried over to the CNV tree. The main difference is that the common, interwoven sections of the trees which correspond to shared history among strains are much shorter in comparison to the strain-unique sections. This suggests a younger history for CNVs compared to SNPs.

SNP- and CNV-based inbred mouse strain phylogenies. Trees generated by the SplitsTree4 hybridization tree-algorithm are shown. Major sections of the SNP-based tree are colored to highlight their locations. This same strain coloring is used for the CNV-based tree. Branch lengths within a tree are proportional to phylogenetic distance, and those with discontinuity markers are actually twice the length shown. Black lines represent regions of hybridization within the trees, where genetic information is being exchanged between lineages or their precursors.
SNP- and CNV-based inbred mouse strain phylogenies. Trees generated by the SplitsTree4 hybridization tree-algorithm are shown. Major sections of the SNP-based tree are colored to highlight their locations. This same strain coloring is used for the CNV-based tree. Branch lengths within a tree are proportional to phylogenetic distance, and those with discontinuity markers are actually twice the length shown. Black lines represent regions of hybridization within the trees, where genetic information is being exchanged between lineages or their precursors.
These results are in line with recent studies which reported ‘high rates of large-scale DNA copy number change’ (Egan et al., 2007) and even CNV heterogeneity within C57BL/6J, which should be a completely inbred strain (Watkins-Chow and Pavan, 2008). In an attempt to calculate the rate of CNV formation in mice from the Cutler data, we selected 11 diverse strain pairs for which breeding history supported a common ancestor strain and with a good estimate for when the strains were created (Beck et al., 2000), including strains from the C57 group, the 129 group, the DBA group, and the FVB/SJL/SWB group. The ratio of CNV differences between the strains versus the number of years since the strain pairs have been separate was calculated for each strain pair (Fig. 4). These values fall within a reasonably narrow range of 0.17 to 0.56 CNVs/year of separation, with a mean of 0.37 CNVs/year. Using, as a very rough estimate, three generations per year (Egan et al., 2007), and taking into account that CNVs accumulate in both branches being compared (thus doubling the effective separation time), we can estimate approximately 0.6 new CNVs per mouse. This should be treated as a lower bound for several reasons, including the fact that we can only capture CNVs which are mappable on the C57BL/6 assembly, which are of sufficient size to detect above background given the aCGH array probe coverage, and which are not missed due to cycles of gain and loss or independent recurrence (Egan et al., 2007).
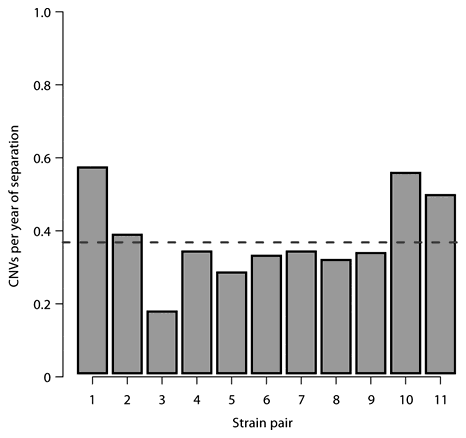
The rate of CNV accumulation between related strain pairs. The ratio of the number of CNVs which vary between a given pair of strains and the number of years of separation of that pair are shown for 11 inbred mouse strain pairs. The mean across these values (0.37 CNVs/year) is indicated by a dashed line. The strain pairs used are: 1) 129S1/SvImJ and 129X1/SvJ (55 years separation); 2) C3H/HeJ and CBA/J (88 years); 3) C57BL/6J and C57BL/10J (71 years); 4) C57BR/cdJ and C57L/J (75 years); 5) C57L/J and C57BL/6J (87 years); 6) C57L/J and C57BL/10J (87 years); 7) C57BR/cdJ and C57BL/6J (87 years); 8) C57BR/cdJ and C57BL/10J (87 years); 9) DBA/1J and DBA/2J (79 years); 10) FVB/Ntac and SWR/J (82 years); 11) FVB/Ntac and SJL/J (82 years).
The rate of CNV accumulation between related strain pairs. The ratio of the number of CNVs which vary between a given pair of strains and the number of years of separation of that pair are shown for 11 inbred mouse strain pairs. The mean across these values (0.37 CNVs/year) is indicated by a dashed line. The strain pairs used are: 1) 129S1/SvImJ and 129X1/SvJ (55 years separation); 2) C3H/HeJ and CBA/J (88 years); 3) C57BL/6J and C57BL/10J (71 years); 4) C57BR/cdJ and C57L/J (75 years); 5) C57L/J and C57BL/6J (87 years); 6) C57L/J and C57BL/10J (87 years); 7) C57BR/cdJ and C57BL/6J (87 years); 8) C57BR/cdJ and C57BL/10J (87 years); 9) DBA/1J and DBA/2J (79 years); 10) FVB/Ntac and SWR/J (82 years); 11) FVB/Ntac and SJL/J (82 years).
CNVs and disease
While it is expected that no essential genes would be found in homozygous deletion CNVs, genes which have significant effects on mouse phenotypes can and are found in both amplification and deletion CNVs. When murine genes overlapping CNVs were mapped to human homologs and compared to the OMIM database (OMIM 2007), 53 genes associated with human disease were identified (Supplementary Table 1, www.karger.com?doi=10.1159/000184721). Of these, only orthologs to three human disease genes were found in deletion CNVs (Cyp2c66, Abca4, and Serpina1 homologs). The human ortholog of the first of these, CYP2C66, which is deleted in CAST/EiJ, is one of the major enzymes responsible for the metabolism of cerivastatin, and was mutated in a patient that had rhabdomyolysis following treatment of hypercholesterolemia with cerivastatin (Ishikawa et al., 2004). The second gene we find deleted is Abca4 in the JF1/Ms strain, with most of its open reading frame absent. This CNV should lead to abnormal rod morphology in the eye as is seen with the laboratory knockout of this gene in 129S4/SvJae as well as in Stargardt disease, a human disease caused by mutations in ABCA4 (Wang et al., 2007). Finally, we observe deletions of members of the Serpina1 family in AKR/J, LP/J, I/LnJ, RIIIS/J, SPRET/EiJ and Balb/cJ mice. SERPINA1, also known as alpha-1 antitrypsin, is a serine proteinase inhibitor. One major physiological function of alpha-1 antitrypsin is the protection of the lower respiratory tract against proteolytic destruction by human leukocyte elastase (HLE). In humans, a hereditary deficiency of alpha-1 antitrypsin is associated with an increased risk of developing emphysema and chronic obstructive pulmonary disease (Crystal, 1990). AKR/J has been shown to be ‘supersusceptible’ to cigarette smoke- induced emphysema as compared to NZWLac/J, C57BL6/J, A/J, and SJ/L strains (Guerassimov et al., 2004). It is of note that these four strains have a full complement of Serpina1 genes. The AKR/J mouse has been found to have slightly lower lung elastance and slightly higher inflammation than the pallid mouse, a C57BL6/J variant strain with very low level of alpha-1 anti-trypsin (de Santi et al., 1995).
Other deletion CNVs which have potential phenotypic impacts are also seen. For example, RIIIS/J, PWK/PhJ, and WSB/EiJ contain deletions in the Ifi204 gene. This gene or its human homolog IFI16 have been implicated in the development of mononuclear phagocytes (Bourette and Mouchiroud, 2008), modulation of ras activity (Ding and Lengyel, 2008), DNA damage and checkpoint control (Ouchi and Ouchi, 2008), as well as cancer and autoimmune disease (Choubey et al., 2008). The deletion CNVs identified cover a region annotated as intronic, but which overlaps sequenced mRNAs as well as spliced ESTs and therefore likely does contain exons.
The remaining 40 genesassociated with human genetic disorders were all found in regions of copy-number increase as compared to C57BL6/J. Although these CNVs may have phenotypic effects, their presence is less likely to result in overt disease as compared to missing genes. Since this summary only covers CNVs that overlap with protein transcriptional reading frames and ignores those which potentially span transcriptional regulatory regions and non protein-coding transcripts, it is only a starting point in understanding the involvement of CNVs in disease etiology in the mouse.
CNVs and mice as model organisms
As discussed earlier, one notable bias in the overlap between genes and CNVs is the preponderance of CNVs covering genes related to defense against pathogens. Gene copy number in killer cell lectin-like receptor (Klr) genes is highly variable (Anderson et al., 2005; Cutler et al., 2007; Graubert et al., 2007) and can explain differences in susceptibility to cytomegalovirus infection (Lee et al., 2001) as well as response to tumor (Nakamura et al., 1999). A number of mouse strains have complete deletions of multiple members of this gene family (Supplementary Table 1), which are concentrated in a locus on chromosome 6 (Fig. 5). Because this variation may affect the ability of these strains to respond to challenges to the innate immune system, researchers studying these processes should consider evaluating their data in light of this CNV data. Although informative, we know that the aCGH analysis is underestimating the CNV diversity of these genes. When directly sequenced, the Balb/cJ mouse was found to not only have the deletion CNVs noted here, but also the presence of additional genes absent from the C57BL6/J genome and therefore not probed for by the arrays (Anderson et al., 2005). This suggests that sequencing of additional strains could lead to the discovery of additional members of this family.

CNVs at the Klra locus. The log2 values for the aCGH data (points) and the locations of called CNVs (boxes) are shown for C57BL/6J and all 20 strains with CNVs in the region of chromosome 6 from nucleotide 129787139 to 103362567. Amplification CNVs and aCGH values within them are shown in red and deletion CNVs and their data points are shown in green. Also indicated are the 3-point running means of the aCGH data as a black line for each strain. At the bottom, the exon (thick line) and intron (thin line) positions for all the Klra genes in this locus are shown in red.
CNVs at the Klra locus. The log2 values for the aCGH data (points) and the locations of called CNVs (boxes) are shown for C57BL/6J and all 20 strains with CNVs in the region of chromosome 6 from nucleotide 129787139 to 103362567. Amplification CNVs and aCGH values within them are shown in red and deletion CNVs and their data points are shown in green. Also indicated are the 3-point running means of the aCGH data as a black line for each strain. At the bottom, the exon (thick line) and intron (thin line) positions for all the Klra genes in this locus are shown in red.
In many strains (SPRET/EiJ, MOLF/EiJ, RIIIS/J, MSM/Ms, and JF1/Ms), we observe high-level increases in copy numbers of the Itln1 gene compared to C57BL6/J. The copy number is likely to be greater than 20 in the RIIIS/J strain. The protein product of this gene (known by various names including intelectin, lactoferrin receptor, and omentin) has been implicated in binding lactoferrin and iron absorption (Suzuki and Lönnerdal, 2004), associated with obesity (Yang et al., 2006), and involved in response to pathogenic infection in the gut (Wrackmeyer et al., 2006). This gene has a paralog (Itln2) in some mouse strains (Pemberton et al., 2004), but which is not present in C57BL6/J so therefore not on the CGH microarrays used by Cutler and Graubert. Orthologs to both genes, Itln1 and Itln2, are present in humans. Interpretations of murine in vivo studies of either protein, or phenotypes related to known functions of these proteins, must take into account this copy-number information.
A locus containing three Raet1 genes (retinoic acid induced early transcript) shows copy number changes in many strains. These major histocompatibility complex (MHC) class I-related gene products have been shown to bind to the C-type lectin receptor, NKG2D (also known as KLRK1), present on natural killer cells as well as certain subsets of T-cells (Lanier, 2005). Because this interaction is involved in immunosurveillance of tumor cells (Strid et al., 2008), models which involve de novo tumor formation (either spontaneous or induced) can be affected by the absence of some or all of these genes.
The Tlr7 gene (as well as nearby genes) on the most distal part of the X chromosome has been shown to be duplicated in SB/Le mice and confers increased severity of systemic lupus erythematosus (SLE) in male mice. By fluorescence in situ hybridization (FISH) mapping, this duplication has been shown to exist on the Y chromosome explaining the nature of this Y-linked autoimmune accelerator (Yaa) locus (Pisitkun et al., 2006). Analysis of the Cutler CNV data suggests that the FVB/Ntac strain also contains a duplication of this locus. While it is unknown whether this duplication exists on the Y chromosome in FVB/Ntac, it is likely to have the same phenotypic consequences.
Another crucial component of the innate immune response is the defensin family (Selsted and Ouellette, 2005). This family of cysteine-rich peptides is expressed in multiple different tissue types including leukocytes and intestinal crypt cells. In mice, α-defensin expression is absent in leukocytes, but several α-defensins can be expressed in the Paneth cells of intestinal crypts. Additionally, more than 50 β-defensin genes have been found in mice. The host response to microbes in the intestinal tract appears to be a vital aspect of the pathogenesis of inflammatory bowel disease (IBD, reviewed in Wang et al., 2007). It has been shown that copy number polymorphisms are prevalent in the β-defensin genes, and reduced copy numbers predispose people to Crohn’s disease (Fellermann et al., 2006). In nearly half of the strains we examined, copy number variations in several of the β-defensins as well as complete loss of the defensin-related cryptdins were found. Unless similar genes exist in these strains to compensate for the missing functions of these defensin-related cryptdins, one would expect that the lack of antimicrobial peptides expressed in the intestinal crypts would predispose these strains to microbial invasion of the mucosa and IBD. Although it is clear that IBD is a multigenic disease with multiple environmental influences, study of these pathologies should consider the copy number changes seen in our mouse models.
The high degree of variation in genes involved in the innate immune response could be due to the way in which laboratory strains are bred and maintained. Over time, gene duplication coupled with diversification leads to the development and expansion of gene families, such as the killer cell lectin-like receptors and defensins discussed above. Each family member may serve overlapping as well as unique functions compared to the other family members. As laboratory strains are largely protected from immune challenges that they might face in the wild, the selective pressure to maintain the diversity of these families could decrease and individual genes can be lost from populations and then that loss will be fixed through inbreeding.
Conclusion
Inbred mouse strains have been an important model for studying complex biological phenotypes since the beginning of the 20th century. The power of this disease model was greatly enhanced with the sequencing of the mouse genome in 2002 (Mouse Genome Sequencing Consortium, 2002). However, a static genome sequence belies the actual dynamic state of the genome as revealed by recent CNV analyses (Li et al., 2004; Snijders et al., 2005; Cutler et al., 2007; Egan et al., 2007; Graubert et al., 2007; Watkins-Chow and Pavan, 2008). Furthermore, this CNV data points to deficiencies in the published genome sequence, specifically the misassembly of some repeated genomic regions and the likelihood that sequences present in other mouse strains are absent from the sequence based on the canonical C57BL/6 strain. Researchers performing reverse genetics in mice, such as creating knockout or mutant strains, need to be aware of the copy-number status of their gene target. The phenotype of a knockout mouse may be very misleading, for example, when a wild-type repeated copy of the targeted gene still exists. Researchers engaged in forward genetics analyses, such as WGA, can also put this knowledge of the mouse CNV landscape to work. This has been demonstrated recently, where an amplification at the glucagon-like peptide-1 receptor locus was found to be linked to food intake levels across an assortment of mouse strains (Cutler et al., 2007). Finally, since inbred strains, in large part, reflect the genomic diversity of the individual mice that served as founders for the strains, their genomes reflect a snapshot in time of genome evolution. This developing picture of the mouse CNV landscape provides insight into the forces that shape genomes in both mice and humans.
Materials and methods
The data used in the analyses shown here all comes from Cutler et al. (2007), unless otherwise noted. Numbers of disparate CNV loci were calculated by ‘collapsing’ CNVs from all strains which either overlapped by at least 8 kb or where one CNV was less than 8 kb and was entirely contained within a larger CNV. Smoothed fold-change histograms were generated using the R function smooth.spline with 38 degrees of freedom on a histogram with 70 breaks. Mouse phylogenetic trees were generated using the SplitsTree4 program (Huson, 1998). For the CNV-based tree, the collapsed CNV set was used. The CNVs were converted into a pseudo-nucleotide-sequence format based on this conversion key: CNV amplitude ≤–10x → ‘AAA’, ≤–2.5x → ‘AAC’, <–1x → ‘ACC’, ≥10x → ‘TTT’, ≥2.5x → ‘TTC’, >1x → ‘TCC’, no CNV → ‘CCC’. SNPs were also converted into a pseudo-sequence file by extracting the 8,315 SNPs with the greatest strain coverage from the SNP database found at http://phenome.jax.org/pub-cgi/phenome/mpdcgi?rtn=snps/download. Ungenotyped SNPs were replaced with ‘N’ and conflicting SNP genotypes in the downloaded data set were replaced with the appropriate ambiguity codes. These pseudo-sequences were then imported into SplitsTree4 as FASTA files. The settings used were, for the distance metric: ‘Uncorrected_P’ with ‘AverageStates’ for ambiguous positions and ‘Normalize’ set to On; for the network type, ‘HybridizationNetwork’ was selected.
