

Abstract
As more and more substances have been shown in preclinical studies to be capable of preventing damage to the inner ear from exposure to noise, ototoxic drugs, ischemia, infection, inflammation, mechanical trauma and other insults, it is becoming very important to develop feasible and safe methods for the targeted delivery of drugs to specific regions in the inner ear. Recently developed methods for sampling perilymph from the cochlea have overcome major technical problems that have distorted previous pharmacokinetic studies of the ear. These measurements show that drug distribution in perilymph is dominated by passive diffusion, resulting in large gradients along the cochlea when drugs are applied intratympanically. Therefore, in order to direct drugs to specific regions of the ear, a variety of delivery strategies are required. To target drugs to the basal cochlear turn and vestibular system while minimizing exposure of the apical cochlear turns, single one-shot intratympanic applications are effective. To increase the amount of drug reaching the apical cochlear turns, repeated intratympanic injections or controlled-release drug delivery systems, such as biodegradable biopolymers or catheters and pumps, are more effective. However, if the applied substance does not easily pass through the round window membrane, or if a more widespread distribution of drug in the ear is required, then intralabyrinthine injections of the substance may be required. Intralabyrinthine injection procedures, which are currently in development in animals, have not yet been proven safe enough for human use.
Introduction
Local drug delivery to the inner ears of humans was first used more than half a century ago for the treatment of Ménière’s disease with local anesthetics [1,2] and antibiotics [3]. It was popularized in the 1990s as it became accepted that locally applied gentamicin provided an effective treatment for the vestibular symptoms of patients with Ménière’s disease with limited risk to hearing [4,5,6]. In addition to aminoglycosides and anesthetics, a variety of drugs have been applied extracochlearly to the round window niche in humans, including neurotransmitters and neurotransmitter antagonists for tinnitus [7], monoclonal antibodies for autoimmune inner ear disease [8] or apoptosis inhibitors (AM-111) for noise-induced hearing loss [9]. However, glucocorticoids have become the most widely used drugs for local application to the inner ear, and have been given to treat Ménière’s disease [10], idiopathic sudden sensorineural hearing loss [11,12,13], autoimmune inner ear disease [14] and tinnitus [15], even though the evidence supporting their use is rather limited [16,17]. Nevertheless, at present, dosing protocols and the selection of drug delivery systems are almost totally empirically based, and there is still only a limited understanding of the pharmacokinetics of drugs in the ear.
Pharmacokinetics of the Inner Ear
Although the ‘LADME’ scheme was developed to describe the pharmacokinetic processes in the human body following a given dosage regimen, it is helpful to adopt this concept for understanding and investigating the principles of drug movements in the inner ear after local or systemic application. The LADME concept involves liberation, absorption, distribution, metabolism and elimination of drugs (fig. 1). While for whole body pharmacokinetics, the LADME processes are centered on blood circulation, in the ear they are centered on the inner ear fluids.

Pharmacokinetic processes of the inner ear according to the LADME concept, as described for an intratympanic application of a formulated drug. Absorption occurs primarily through the round window membrane. The drug, upon entering the perilymph, distributes both within the scala tympani and into adjacent fluid and tissue-filled spaces. The drug is also subjected to metabolism and elimination to blood or CSF.
Pharmacokinetic processes of the inner ear according to the LADME concept, as described for an intratympanic application of a formulated drug. Absorption occurs primarily through the round window membrane. The drug, upon entering the perilymph, distributes both within the scala tympani and into adjacent fluid and tissue-filled spaces. The drug is also subjected to metabolism and elimination to blood or CSF.
Liberation describes the release of the drug from its dosage form.
Absorption refers to the movement of the drug from the site of administration to the inner ear fluids (e.g. from the middle ear to the perilymph of the scala tympani (ST) through the round window membrane, RWM).
Distribution involves the processes by which the drug diffuses, flows or is transferred within and between the different fluid-filled compartments (perilymph and endolymph), and how it spreads from the fluid spaces into the various tissue compartments of the inner ear.
Metabolism is the chemical conversion or transformation of drugs into active moieties or compounds which are easier to eliminate.
Elimination describes the removal of the unchanged drug or metabolite from the inner ear (e.g. to blood, cerebrospinal fluid or the middle ear).
Although these processes generally follow the above sequence, they may occur simultaneously. While the drug is still being liberated from a controlled release formulation, previously absorbed drug may already have been eliminated.
Liberation
Current efforts in the area of drug delivery in general, and also specifically in inner ear therapies, include the development of drugs which are liberated from a formulation over a period of time in a controlled manner. Types of sustained release formulations include liposomes, drug-loaded biodegradable microspheres and drug polymer conjugates, including gels [18,19,20,21,22]. Drug release from carrier systems may be driven only by the concentration gradient (such as for a drug in a resorbable gelatin sponge soaked with drug solution) or maintained by a gradual breakdown of the carrier, either spontaneously or induced by physical and chemical triggers (e.g. temperature or pH), with subsequent release of drug.
Absorption
Absorption of a drug from the middle ear to perilymph of the inner ear can occur through a number of structures, including:
Round Window Membrane. The RWM in humans, monkeys, felines and rodents consists of 3 main layers: (1) an outer epithelial layer facing the middle ear cavity; (2) a middle connective tissue layer; (3) an inner cellular layer facing the ST perilymph [23,24]. Tight junctions are present between cells of the outer layer, while in the middle layer fibroblasts, fibrocytes, collagen, elastin, capillaries, and myelinated and unmyelinated nerves have been described [25]. Many studies have demonstrated in qualitative terms that substances applied to the middle ear enter the basal turn of the ST, and may influence structure and function of the ear [24,26,27,28]. In contrast, few have performed quantitative measurements of drug levels in the perilymph or measured RWM permeability. Of the pharmacokinetic studies in the literature, a substantial proportion cannot be interpreted quantitatively due to sampling methods that caused the fluid samples to be highly contaminated with CSF [18,29,30,31]. In these studies, large volumes (10 µl) relative to the volume of the ST in the guinea pig (4.6 µl) [32] were taken from the basal turn of ST. As the cochlear aqueduct enters ST at this location, samples taken nearby become severely contaminated with CSF that is drawn into the scala as the sample is aspirated. Based on measurements with marker ions, it was estimated that 10-µl samples taken from the basal turn of guinea pigs contained as little as 15% perilymph and 85% CSF [33]. Sample measurements are more readily interpreted when the samples are taken from a location further from the cochlear aqueduct. A better technique, in which multiple samples are taken sequentially from the cochlear apex within a period of a few minutes, allows both the concentration and the gradient of drug along the ST to be quantified [34]. Results obtained with this technique show that, following 2- to 3-hour application of a drug or marker to the RWM, there are substantial gradients along the ST. The gradients determined for 3 substances are shown in figure 2. For TMPA (trimethylphenylammonium: an ionic marker), gentamicin and dexamethasone, basal-apical concentration differences of over 1000-fold were found in most animals. The presence of basal-apical gradients following drug applications to the RWM is supported by a number of histological studies that suggested markers were at higher concentration or cellular damage was greater in the basal turn than in apical regions [28,35,36]. The concentration measurements in figure 2 also show that the basal turn concentration of drugs is variable, with over 10-fold differences between animals being common. Measurements of entry rates using microdialysis have confirmed that the variability between animals arises from differences in RWM permeability [37]. RWM permeability has also been shown to be sensitive to experimental manipulations. Permeability is increased by local anesthetics [38], endotoxins and exotoxins [39,40], histamine [41], drying through the use of suction near the round window niche [42], by osmotic disturbances and by the presence of benzyl alcohol (a commonly used preservative) in the applied solution [42].
![Fig. 2. Concentration gradients along ST of the guinea pig following 2- to 3-hour applications to the RWM. Distances are measured along the ST from the basal end. Results are shown for the marker ion TMPA [34], gentamicin [53] and dexamethasone [54]. In addition to the steep concentration gradients, it is also apparent that the basal turn (0–2 mm) concentrations of each substance vary by more than a factor of 10, due to inter-animal variations in RWM permeability and elimination of the drug. Recently, it has been shown that these concentration gradients are stable with time [83].](https://karger.silverchair-cdn.com/karger/content_public/journal/aud/14/6/10.1159_000241892/2/m_000241892_f02.gif?Expires=1716298412&Signature=AaZU1pHGYh8rl7YeUmZUJuT31QAD-l-5bjQf0HZYzqkG0dkt~hCf37oLR5EmKSXRfxgKt4EX9RCx5s3Pr6YZsCRKTNWMTkyquocsIvne2j4OqLn4Wim0IAJJ59xjx17TbsKCrez61fZ8cmHhM6QrVLugdrwdlYKwWn4246bNUqgMQOscTEFu2S5-WoZuZvZgq7HtDvwtLFlmo-tSFy9eowtYxP~M1QMkbfMw4XamvwB8qr6LwtMEjqsfWJmZ5ImOMRT2a~d2CRjw7fz6pHxoftWUcXNdc2Grk5HkfWWd4z-Qh4YxPf6wNQChjAm9cSmNdk6zWox~iA7qeD2iXltXJw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Concentration gradients along ST of the guinea pig following 2- to 3-hour applications to the RWM. Distances are measured along the ST from the basal end. Results are shown for the marker ion TMPA [34], gentamicin [53] and dexamethasone [54]. In addition to the steep concentration gradients, it is also apparent that the basal turn (0–2 mm) concentrations of each substance vary by more than a factor of 10, due to inter-animal variations in RWM permeability and elimination of the drug. Recently, it has been shown that these concentration gradients are stable with time [83].
Concentration gradients along ST of the guinea pig following 2- to 3-hour applications to the RWM. Distances are measured along the ST from the basal end. Results are shown for the marker ion TMPA [34], gentamicin [53] and dexamethasone [54]. In addition to the steep concentration gradients, it is also apparent that the basal turn (0–2 mm) concentrations of each substance vary by more than a factor of 10, due to inter-animal variations in RWM permeability and elimination of the drug. Recently, it has been shown that these concentration gradients are stable with time [83].
In addition, there is only limited knowledge about the individual processes of transmembrane transport contributing to substance absorption through the RWM, including passive diffusion, facilitated diffusion through carriers, active transport or phagocytosis.
Oval Window (Including the Stapes Footplate and Annular Ligament). Although a number of investigators have suggested substances may enter perilymph of the vestibule by this route [36,43], it is technically difficult to measure the amount. Substances cross readily between the ST and scala vestibuli (SV) [44,45], presumably passing through the spiral ligament (discussed further in ‘Distribution’). Thus, the observation of drug or marker in the vestibule or saccule does not confirm that it entered through the oval window. In addition, attempts to occlude the RWM (such as with dental cement [36]) were found to be only partially effective. It is undoubtedly possible for drugs to pass through the thin bone of the stapes footplate or through the walls of the oval window niche in amounts which may be significant in the human. The amount of drug entering by this route, however, remains uncertain and likely depends on drug size and charge, but is thought to be small relative to that entering through the RWM.
The Bony Otic Capsule. It has recently been shown that when a drug is applied by filling the middle ear with solution in guinea pigs, the highest drug levels are produced in the apical regions of the cochlea [46]. This results from the drug entering perilymph through the bony otic capsule, which is very thin in the apical turns of animals such as guinea pigs and chinchillas. This presents a considerable problem when studies in rodents are used as a model for drug delivery in the human, as it can be assumed that the thicker bone of the human otic capsule will represent a more effective boundary. The resulting drug distribution patterns along the length of the cochlea are therefore likely to differ markedly between animals and humans following intratympanic applications.
Distribution
The fluids of the inner ear show little evidence of ‘stirring’, i.e. no pronounced movement comparable to that of the systemic circulation. In the intact state, rates of volume flow of perilymph and endolymph are both exceedingly slow and the distribution of drugs within the fluid spaces is dominated by passive diffusion. Diffusion is a highly predictable process and its effects can be calculated with accuracy. The rate of substance movement by diffusion is nonlinear with distance, allowing drugs to spread rapidly over short distances (such as across a cochlear scala), but slowly over distances of more than a few millimeters. This results in large gradients along the cochlea when substances are applied to the basal turn, as shown earlier in figure 2. Also contributing to the gradients along the length of the cochlea is the loss of drug from the scala as it diffuses. Losses result from the distribution into fluid spaces and tissue compartments adjacent to the scala, and through elimination from the ear to other much larger compartments, such as blood. For ST, substances readily distribute through the fluid spaces of the spiral ligament into the SV and the vestibule. Figure 3 shows the anatomy of the spiral ligament in the basal turn, which is seen to wrap around the periphery of approximately half the RWM. Drugs present in the perilymph at the base of ST close to the RWM would therefore be expected to have ready access to perilymph of the vestibule. In addition, drugs in the ST can pass through passages in the bone (canaliculi perforantes) into Rosenthal’s canal and from there to the modiolus [47,48], and into the fluid spaces of the organ of Corti [49]. Although the canaliculi perforantes in the medial wall of the ST appear rather large, data from experiments with neurotrophins suggest that only a small proportion of a drug infused into the ST reaches the vicinity of the spiral ganglion neuron cell bodies, suggesting that other barriers to diffusion may exist [50].
![Fig. 3. Reconstructed 3D anatomy of the fluid spaces of the guinea pig inner ear derived by segmentation of an OPFOS (orthogonal-plane fluorescence optical sectioning) image set [84] using Amira software. The enlargement shows the basal turn with the stapes removed (leaving an imprint of the footplate). V = Vestibule; SL = spiral ligament; RW = round window. The SL follows the periphery of the RW almost half way around, providing a major route for drugs in the ST near the RW to diffuse across into the vestibule. This anatomic pathway accounts for how drugs applied intratympanically to the RW can gain access to vestibular structures. Blue = Endolymph; orange = perilymph of SV and V; yellow = perilymph of ST; green = spiral ligament; red = sensory structures; purple = RW; magenta = cochlear aqueduct; brown = stapes.](https://karger.silverchair-cdn.com/karger/content_public/journal/aud/14/6/10.1159_000241892/2/m_000241892_f03.jpeg?Expires=1716298412&Signature=1FVZT0ZCvAnvQ~ixrK4sLz7xoukW-hy-unj359Jd6Z~K7Z5SuP6Fa8vNJEDZtOIyt0tv6LSfW6V2PYp2BvLMb2p6ZXKOfaJlJJMWDESHDU8KQw120i~-vuPBhlCnH0DGGii--sz4R-c-ZscD8C~wFzz4chqiOsnsMfHcmBb6W~GCcdSi7tSEtMnEEoYvYTScuOLDvRgmtlrUcQ-x3DEHDmFxk52ehjyioDi0LlxKQticoQQt4MfNeawH4ip61cjtefQe~3fqsuwI33XSQuhNttlsDRMkXaaSNtRoIPx3wawmKUmledkP1bKjffsPKoOnTroIVvJld8UMqPgbA9d-XQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Reconstructed 3D anatomy of the fluid spaces of the guinea pig inner ear derived by segmentation of an OPFOS (orthogonal-plane fluorescence optical sectioning) image set [84] using Amira software. The enlargement shows the basal turn with the stapes removed (leaving an imprint of the footplate). V = Vestibule; SL = spiral ligament; RW = round window. The SL follows the periphery of the RW almost half way around, providing a major route for drugs in the ST near the RW to diffuse across into the vestibule. This anatomic pathway accounts for how drugs applied intratympanically to the RW can gain access to vestibular structures. Blue = Endolymph; orange = perilymph of SV and V; yellow = perilymph of ST; green = spiral ligament; red = sensory structures; purple = RW; magenta = cochlear aqueduct; brown = stapes.
Reconstructed 3D anatomy of the fluid spaces of the guinea pig inner ear derived by segmentation of an OPFOS (orthogonal-plane fluorescence optical sectioning) image set [84] using Amira software. The enlargement shows the basal turn with the stapes removed (leaving an imprint of the footplate). V = Vestibule; SL = spiral ligament; RW = round window. The SL follows the periphery of the RW almost half way around, providing a major route for drugs in the ST near the RW to diffuse across into the vestibule. This anatomic pathway accounts for how drugs applied intratympanically to the RW can gain access to vestibular structures. Blue = Endolymph; orange = perilymph of SV and V; yellow = perilymph of ST; green = spiral ligament; red = sensory structures; purple = RW; magenta = cochlear aqueduct; brown = stapes.
In addition, some substances may also enter endolymph, although this is likely to depend on the electrical charge of the molecule, which subjects it to the influence of cellular potentials and the endocochlear potential. Cationic markers are excluded from endolymph [44], while anionic markers are accumulated and retained there [51].
Apart from a drug’s chemical characteristics, especially size and its hydrophobicity, distribution processes are primarily dependent on concentration gradients, so that the distribution of substances into different compartments of the inner ear can be reversed if the concentration gradient changes. This is apparent during sequential apical sampling, when CSF entering the basal turn of the ST gains drug as it passes through the scala towards the sampling site, and can be referred to as ‘redistribution’. Indeed, the rate at which sequential sample concentrations decline during rapidly repeated sampling from the apex provides a valuable index of the rate and amount of drug available to redistribute into the scala during sampling.
It has also been suggested that protein levels in perilymph may be sufficient to bind or buffer some drugs [52]. Uptake of a drug into cellular structures of the ear may also occur. By causing a loss of drug from the scala, each of these processes acts to reduce the rate at which the drug spreads along the scala by diffusion. However, in contrast to the expected slowing of the rate of distribution caused by these processes, the measured gradients along the ST in figure 2 could only be explained by the substances spreading towards the apex at a slightly faster rate than can be accounted for by diffusion. To account for the data, it was necessary to incorporate a low rate of apically directed perilymph flow into the simulations of the experiments. The perilymph flow rates averaged 0.019, 0.021 and 0.009 µl/min for TMPA [34], gentamicin [53] and dexamethasone [54], respectively. While these rates are extremely low, they suggest that perilymph flow may contribute significantly to drug distribution when its effects are accumulated over a period of hours or days.
Metabolism
Metabolism or biotransformation considers the transformation of the drug from one form to another with altered (lower or higher) effectiveness by tissues and enzymes within the ear or in adjacent compartments, such as the middle ear. There is, however, very limited quantitative data about the metabolism of specific groups of substances following their application to the inner ear. It has been shown that dexamethasone-phosphate is metabolized within the ear to the active moiety dexamethasone [54,55]. There is also accumulating evidence that extracellular purines in the cochlear fluids, important modulators of cochlear function and potential targets for manipulation [56], are regulated by ectonucleotidases [57].
Elimination
The rate of elimination of a drug from perilymph is one of the major factors influencing both the concentration achieved in perilymph and how far towards the apex a drug is distributed following its application to the RWM. For faster elimination rates, perilymph concentration saturates at a lower value and reaches a steady state more quickly than with slower elimination rates. In addition, when the amount of drug eliminated from a specific region becomes equal to the rate of diffusion along the scala at that point, then a steady state will be established and the drug will never reach a higher concentration at more apical locations. In order to determine how far a specific drug will spread along the cochlea, knowledge of the elimination rate is required. Determination of the elimination rate for a specific substance is, however, technically difficult. If drug is applied locally to the RWM, then the decline in concentration at a location near the application site (e.g. in the basal part of the ST) following the application results from the combined effects of both distribution and elimination, which would distort quantification of the elimination rate. If the time course of drug decline is monitored by taking samples repeatedly over a long period, such as samples taken hourly in the study by Parnes et al. [29], then the act of sampling, accompanied by drawing CSF into the cochlea, also contributes to the decline of drug concentration and prevents an accurate estimation of the elimination rate. Similarly, the use of microdialysis to follow perilymph concentration as a function of time [37,58] does not provide a valid indication of elimination rate as the microdialysis procedure itself provides a major source of elimination [37]. As a result of these technical difficulties, there are relatively few studies from which drug elimination rates can be reliably derived. In a few studies, where all or most of the perilymph in the cochlea was taken as a single sample, the effects of redistribution were minimized. One series of studies investigated the kinetics of gentamicin in the chinchilla [59], in which large samples of perilymph (15 µl) were taken from the vestibule. These studies were analyzed quantitatively [60] from which a perilymph elimination half-time of 500 min for gentamicin was determined. In another study, prednisolone kinetics were investigated in guinea pigs [61], obtaining perilymph by aspirating the entire contents of the ST and SV after the temporal bone was removed from the animal. Quantitative analysis of these data allowed an elimination half-life of 130 min for prednisolone to be derived [62]. These appear to be the only studies in which the experimental design allowed a meaningful interpretation of the elimination rate for drugs.
While pharmacokinetic analyses generally consider elimination of solutes from the scala fluids to blood, we know from anatomic data that this is an oversimplification. The major capillary beds in the ear are not located in, or directly associated with, the scalae. Although arterioles and venules run around the bony walls of SV and ST, respectively, they run within bony canals separated from the scala fluid spaces. Instead, the major capillary beds are associated with the spiral ganglion, the lateral wall and limited regions of the organ of Corti. Fluid pathways through the bone between the ST and the spiral ganglion, and between the SV and the modiolar spaces, have been demonstrated [47,48]. Elimination of drugs from perilymph is therefore likely to occur indirectly, mediated by the vascularized tissues in communication with perilymph. Whether spiral ligament or modiolar sites dominate perilymph kinetics remains to be established. It is also likely that distribution of many substances will be influenced by active transport processes at physiological boundaries in the ear, comparable to those established for the eye [63], but given the complexities of pharmacokinetic studies of the ear, influences of specific transport processes on cochlear fluid pharmacokinetics have yet to be demonstrated.
Delivery Systems and Protocols
Based on knowledge of how drugs enter and are dispersed in the ear, it is possible to build a framework for how drugs can be delivered to the ear for specific purposes, as shown in figure 4. At present, the strategy for local drug delivery in clinical situations is dominated by intratympanic applications. For situations where the drug does not readily pass the RWM, or where better control of drug level is necessary, then intralabyrinthine applications are necessary. However, none of the intralabyrinthine application techniques used in animals has yet been proven safe enough for use in humans, although this field is advancing rapidly.
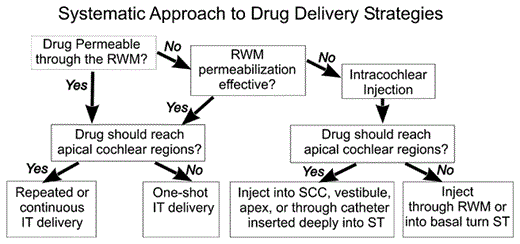
Idealized flowchart for drug delivery to the ear, taking into account the known distribution properties and limitations of different delivery systems. IT = Intratympanic; SCC = semi-circular canal.
Idealized flowchart for drug delivery to the ear, taking into account the known distribution properties and limitations of different delivery systems. IT = Intratympanic; SCC = semi-circular canal.
Intratympanic (Middle Ear) Applications
Delivery protocols used in conjunction with intratympanic applications have included single injections (a ‘one-shot’), repeated injections (3 times a day over a period of time, or longer intervals such as weekly), or continuous delivery over a period of days using a microcatheter. The influence of delivery protocol on the amount and time course of drug in perilymph has not yet been documented, but calculations show that a key variable influencing the perilymph concentration achieved by each application is the time the drug remains in contact with the RWM [37]. As most of the decline in drug level near the application site results from distribution of the drug into other parts of the ear, longer application times help maintain the drug level while nearby regions become loaded with the drug. In clinical terms, this means that to reduce variability of perilymph drug levels to a minimum, the time the drug remains in the round window niche must be controlled as closely as possible. Calculations also show that multiple injection or continuous application protocols not only produce higher drug concentrations near the application site, but also allow proportionately greater concentrations to reach higher turns of the cochlea [64]. The application protocol can therefore be modified according to the goals of the local drug application. For treatment of Ménière’s patients with gentamicin, where the goal is to suppress the balance system while minimizing gentamicin-induced hearing loss, a one-shot application is most appropriate, as this results in the steepest drug gradient along the cochlea, minimizing the gentamicin concentration reaching cochlear locations responsible for speech frequencies. In contrast, when the goal is to distribute the drug throughout the cochlea, then multiple applications per day or continuous delivery protocols would minimize longitudinal gradients to the greatest degree possible. There are numerous other factors that influence the perilymph drug level resulting from a specific application protocol, including the RWM permeability to the drug and how fast the drug is eliminated from the round window niche by the middle ear mucosa, through drainage to the spaces of the temporal bone, and by the injected solution leaving the middle ear through the eustachian tube.
Intralabyrinthine Applications
In the literature, the ambiguous terms ‘infusion’, ‘perfusion’ and ‘injection’ have been used to describe the delivery of drugs to the inner ear. The term ‘perfusion’ of the ear was originally used to describe the passage of solution through the perilymphatic spaces from a site of entry to an open outlet. Perfusion was, however, also used to describe intratympanic application without perforation of the otic capsule, i.e. the solution passing solution across the ear [14,65]. ‘Infusion’ (derived from the Latin verb ‘to pour into’) also suggests a passive slow delivery of solution to the ear. In contrast, the terms ‘intralabyrinthine injection’ and ‘intracochlear injection’ are preferred as they more accurately represent a more rapidly occurring and forceful introduction of solution directly into the fluid spaces of the inner ear.
In general pharmacokinetics, the term bioavailability is commonly used to describe the rate and extent of drug input. For extracochlear applications, such as intratympanic injections to the round window niche, the percentage of drug entering the inner ear (bioavailability) is relatively low. It was estimated that the basal turn concentration only reached a mean of 2.5% of the applied gentamicin concentration and 1.4% of the applied dexamethasone-phosphate concentration with a round window irrigation protocol and 0.17% of the applied gentamicin concentration when the bulla was filled with solution [46,53]. Drugs injected directly into the perilymph would theoretically be expected to exhibit 100% bioavailability, and in this respect are comparable to an intravenous application when considering the entire human body. In reality, 100% bioavailability might not be reached in all intralabyrinthine application techniques due to leaks either at the application site or at sites of fluid efflux, such as the cochlear aqueduct. Nevertheless, bioavailability of drug within the inner ear can be substantially increased by direct application of the drug through the RWM, through the stapes footplate, into the lateral semicircular canals or into the endolymphatic sac.
Under conditions when the injection pipette is sealed into the otic capsule, intralabyrinthine injections produced more consistent perilymph concentrations (of the marker ion) than applications to the round window niche [66]. Difficulties arise, however, when there is any form of leak at the injection site or when an outlet for the injected solution is provided. For the same reasons that perforations of the otic capsule cause contamination of fluid samples with CSF, perforations to insert an injection pipette cause a release of intracochlear pressure that results in an artifactual volume flow from the cochlear aqueduct entering the ST in the basal turn to the site of the perforation. In guinea pigs, CSF entry rates of 0.5–1 µl/min occur, which is fast enough to displace drug solution from the ear (especially from the basal turn of ST, where CSF enters) within minutes. Attempts to seal injection sites through the bone with bone dust, adipose tissue or fascia may be effective after a number of hours or days as the site scars, but are unlikely to provide an immediate fluid-tight seal. Thus, the procedures used to seal the injection site may critically influence the perilymph drug level. In humans, the situation may be considerably different from animal models as the cochlear aqueduct is narrower and CSF pressure at the level of the aqueduct may be negative when the patient is sitting or standing.
The spread of drug from the site of injection depends on the rate of injection and the site (if any) where the displaced perilymph exits the ear. For low injection rates, the induced rate of perilymph flow may be low, and spread from the injection site will be dominated by diffusion. This has been demonstrated both by real-time measurements of markers [67] and by functional measures during pulsed injections into the basal turn of the ST [68]. For higher injection rates, the spread of drug can only be predicted if the fluid outlet site is known. As a result of these factors, the region(s) of the inner ear affected by intralabyrinthine injections depends to a large extent on the site of injection. Possible injection sites (as shown in fig. 4) include the RWM (through fenestrations in the bone in the basal turn of ST), the stapes footplate or the semi-circular canals, and the endolymphatic sac.
Many studies have reported the injection of agents through the RWM using hand-held narrow-gauge hypodermic syringes. However, when perilymph concentration was measured following injections through the RWM with fine (20-µM OD) bevelled pipettes held in a micromanipulator, small leaks around the pipettes caused significant washout of the drug [67]. It was calculated that the leakage rate under these conditions averaged just 90 nl/min, which was sufficient to wash out the drug but was not visible with an operating microscope. The leakage around the pipette was markedly reduced when the round window niche was filled with Healon gel before the injection pipette was inserted. These results suggest that injections through the RWM with handheld hypodermic syringes are likely to result in even higher rates of perilymph leakage, and cannot be regarded as a quantitative delivery method.
Cochlear Implants
There is accumulating evidence from animal experiments that locally applied glucocorticoids can help preserve acoustic hearing thresholds following cochlear implantation [69,70,71]. As there is increasing interest in the possibility of implanting patients with high-frequency hearing loss but good low-frequency hearing, it has become an important goal to perform the implantation with minimal damage to residual hearing [72,73,74]. Other drug candidates include the use of neurotrophins to preserve spiral ganglion cells [75] and apoptosis inhibitors to minimize insertion trauma [76]. There are several possible strategies of intracochlear drug delivery in combination with cochlear implants, including applications to the RWM prior to surgery [77], one-shot injections into the ST at the time of implantation [78,79], ‘bathing’ the electrode in drug solution or a gel preparation prior to insertion into the cochlea, drug release from the electrode carrier itself which also functions as a scaffold, drug release from a reservoir in the electrode carrier, drug injection through an incorporated channel attached to a pump [80,81], or by surface coating of the electrode carrier with a controlled-release formulation [82].
Clinical Considerations
At present, the most widely used approach to deliver drugs locally to the inner ear is through the use of intratympanic applications. This is in large part because of the requirement for a safe and feasible way of drug delivery to the inner ear, especially to minimize damage to hearing, which became a major component of local gentamicin applications in the treatment of Ménière’s disease. Animal studies, however, showed that perilymph drug levels achieved with RWM applications are highly variable [29,37,54,61] and that more consistent perilymph concentrations can be achieved by intracochlear injections of substances [67]. However, based on experience with surgical procedures that involve perforation of the inner ear such as stapedectomy or the cochlear implantation of patients with residual hearing, it is well known that perforation and/or surgical manipulation of the ear carries a significant risk of deafness. Appropriate techniques for the intralabyrinthine injections of drugs have not yet been developed and proven safe. This results in a balance or trade-off between the ability of the delivery method to control the perilymph drug level and the risk of the drug delivery procedure to the hearing and balance function of the patient as schematized in figure 5. In order to have good control of perilymph drug levels and effective drug delivery without damage to the ear, we either need to reduce the variability and increase the bioavailability associated with intratympanic applications (such as by better control of the drug level in the round window niche or by permeabilizing the RWM) or we need to develop safe methods of intracochlear drug delivery that do not damage the ear.

Schematic of the balance between the control of perilymph drug level by different application approaches and the risk to the patient’s hearing and/or balance by the drug application alone. Intratympanic drug applications are low risk, but produce variable perilymph levels. Improved control of drug levels may be achieved by manipulations of RWM permeability (arrows). Intracochlear drug injections give better control of perilymph drug concentration, but the safety of such procedures has not been demonstrated. Delivering drugs from a cochlear implant may carry little additional risk to the patient. As experience is gained, the goal remains to provide quantitative control of the drug level in perilymph while minimizing risk to hearing and balance function.
Schematic of the balance between the control of perilymph drug level by different application approaches and the risk to the patient’s hearing and/or balance by the drug application alone. Intratympanic drug applications are low risk, but produce variable perilymph levels. Improved control of drug levels may be achieved by manipulations of RWM permeability (arrows). Intracochlear drug injections give better control of perilymph drug concentration, but the safety of such procedures has not been demonstrated. Delivering drugs from a cochlear implant may carry little additional risk to the patient. As experience is gained, the goal remains to provide quantitative control of the drug level in perilymph while minimizing risk to hearing and balance function.
Acknowledgements
This study was supported by research grant RO1 DC01368 (to A.N.S.) from the National Institute on Deafness and other Communication Disorders (NIDCD), National Institutes of Health (NIH), and by grants 0313844B and 0314103 (to S.K.P.) from the Federal Ministry of Education and Research (BMBF), Germany.
References
A.N.S. and S.K.P. contributed equally to this paper. A.N.S. is a member of the scientific advisory board of Otonomy; however, this work was not supported by Otonomy.
