

Abstract
Background: Serum-derived hyaluronan (HA)-associated proteins (SHAPs), the heavy chains of inter-α-trypsin inhibitor, covalently bind to HA to form the SHAP-HA complex. The SHAP-HA complex is involved in the pathophysiology of inflammatory diseases, including rheumatoid arthritis. We investigated whether this complex is also involved in airway allergy. Methods: SHAP-HA-deficient (bikunin knockout, KO) mice and wild-type (WT) mice were immunized twice by intraperitoneal injection of ovalbumin (OVA) and exposed to aerosol OVA for 30 min each day for 2 weeks. Twenty-four hours after the final OVA challenge, airway responsiveness to inhaled methacholine (MCh) was measured, and analysis of bronchoalveolar lavage fluid (BALF) and lung histological studies were done. Results: Compared to WT mice, KO mice showed higher airway hyperresponsiveness to inhaled MCh and higher late-phase responses to OVA whereas the early-phase responses were similar. Cell differentials of BALF showed an increased number of macrophages and neutrophils in KO mice. Furthermore, decreased concentrations of soluble tumor necrosis factor receptor-1 (sTNFR1) were found in BALF from KO mice whereas the levels of Th1 and Th2 cytokines were not different from WT mice. Immunochemical study of the lung tissues revealed stronger staining of sTNFR1 in KO than in WT mice. Conclusions: Our results suggest that in this murine asthma model, the SHAP-HA complex has an inhibitory role in the development of airway hyperresponsiveness and allergic airway inflammation which may be attributed, at least in part, to negative feedback mechanisms exerted by sTNFR1, the shedding of which from the cell surface might also be promoted by the SHAP-HA complex.
Introduction
Bronchial asthma is characterized by airway allergic inflammation, airway hyperresponsiveness to various stimuli (such as histamine, allergens, viruses, pollutants and cholinergic agonists) and airway remodeling. The remodeling of airways consists of abnormal invasion by inflammatory cells (eosinophils, lymphocytes, macrophages) and myofibroblasts [1,2,3,4], excessive proliferation of airway smooth muscle cells [5] and, consequently, deposition of a hyaluronan (HA)-rich extracellular matrix in the bronchial mucosa [6,7,8].
Studies on the functions of HA in respiratory disease revealed that it is either proinflammatory or anti-inflammatory depending on the processes [7,9]. For instance, HA suppresses elastase secretion by neutrophils and macrophages in pulmonary emphysema [10], and reduces exercise-induced nonspecific bronchoprovocation [11,12]. On the other hand, increased amounts of HA were found in asthmatic airway mucosa and bronchoalveolar lavage fluid (BALF), suggesting that HA may contribute to the pathophysiology of asthma [6,13]. In a murine model of bleomycin-induced lung injury, HA fragmentation was a major response in lung inflammation [14]. The versatility of HA functions may be attributed to its unique physiochemical and biological properties, which largely depend on its concentration, its chain length, and the large number of HA-binding proteins (HABPs) that interact with it. Among the long list of identified HABPs, we identified the only known protein that is covalently bound to HA, serum-derived HA-associated protein (SHAP), which is identical to the heavy chains of inter-α-trypsin inhibitor (IαI) [15,16,17]. IαI, a relatively abundant serum macromolecule, is composed of a light chain (called bikunin, the trypsin inhibitor), which has a chondroitin sulfate chain. Two heavy chains are covalently bound by ester linkages to N-acetylgalactosamine residues in the chondroitin sulfate chain. The SHAP-HA complexes form via an enzyme-catalyzed trans-esterification reaction in which heavy chains are transferred from the chondroitin sulfate chains to N-acetylglucosamine residues in HA. In the SHAP-HA-deficient bikunin-knockout mouse (KO mouse), the heavy chains are present in the circulation in a precursor form, but are not able to form a covalent complex with HA [18].
The formation of the SHAP-HA complex is associated with many inflammatory diseases; large amounts of the SHAP-HA are notably found in the synovial fluid and hyperplasic synovium of patients with rheumatoid arthritis [19] and in the hyperplastic muscularis mucosae [20]. Further, the SHAP-HA complex enhances CD44-mediated leukocyte adhesion [19], and promotes angiogenesis in fibrotic lung injury [21].
In the present study, we explored the role of the SHAP-HA complex in the pathogenesis of airway hyperresponsiveness in an ovalbumin (OVA)-induced murine asthma model using KO mice and wild-type (WT) mice. The results demonstrate that the SHAP-HA complex stabilized the HA-rich extracellular matrix and likely suppressed the asthmatic inflammation by inhibiting tumor necrosis factor (TNF)-α activity through interaction with soluble TNF receptor-1 (sTNFR1).
Materials and Methods
Animals
Bikunin-KO mice cannot form IαI, which is required to form SHAP-HA complexes [18], and are therefore considered as SHAP-HA complex-deficient mice. Bikunin heterozygous (bikunin+/–) mice in a BALB/c genetic background (at the 9th generation of backcrossing) were mated to produce KO (bikunin–/–) and WT (bikunin+/+) mice. Seven-week-old female bikunin–/– and bikunin+/+ mice were chosen for this study. All mice were maintained in a specific pathogen-free environment with a 12-hour/12-hour light-dark cycle, and were provided with an OVA-free diet and water. All experiments were conducted with the approval of the University Animal Care and Use Committee.
Sensitization and Airway Challenge
Mice were sensitized on day 0 and on day 13 by intraperitoneal injections of 50 µg of OVA grade V (Sigma, St. Louis, Mo., USA) and 1 mg aluminum hydroxide (Sigma) in a total volume of 0.5 ml. From day 23, mice were exposed to aerosolized OVA (1% in sterile saline) for 30 min per day during the following 2 weeks (fig. 1). Control animals were WT mice and were treated in the same manner by injecting them with saline plus aluminum hydroxide and exposing them to saline aerosol.

Measurement of Airway Responses
Mice were assessed for their airway responses to methacholine (MCh) (Sigma) by using noninvasive whole-body plethysmography (Buxco Eletronics Inc., Troy, N.Y., USA) 24 h before and on day 37, after the 2-week airway challenge (fig. 1), respectively. Mice were exposed to aerosolized MCh for 3 min and immediately put into the main chamber, followed by recording their inspiratory/expiratory flow during unrestrained conscious respiration for 3 min. The process was repeated with MCh at increasing concentrations. The enhanced-pause (Penh) values were calculated and used as an indicator of airway response [22,23].
Airway response to OVA was also measured on day 38 in the same way as described above. Inspiratory/expiratory flow was recorded before, immediately after, and 8 h after the exposure to nebulized OVA (1% in sterile saline) for 30 min to calculate the baseline Penh value, the early-phase response (EPR), and the late-phase response (LPR). In this study, every Penh value was averaged for 3 min.
Bronchoalveolar Lavage and Differential Cell Count
On day 39, mice were injected intraperitoneally with a lethal dose of sodium pentobarbitone (200 mg/kg). After blood collection from the inferior vena cava, the trachea was exposed, and the left bronchus was tied to keep the left lung unlavaged for histological examination. The right lung was lavaged three times with 0.5 ml each of Hanks’ balanced salt solution via a polyethylene catheter (1.1 × 30 mm, Becton Dickinson Infusion Therapy System Inc., Sandy, Utah, USA). After counting total cells in BALF with a hemocytometer, cytospin slides were made and stained with Diff-Quick (IMEB, Japan). The slides were examined for differential cell count in a blinded fashion by counting at least 300 cells under a light microscope.
Measurement of Antigen-Specific IgE and IgG1 Levels
Serum levels of OVA-specific IgE and IgG1 were measured with an ELISA kit (AKRIE Corp., Shibayagi, Gunma, Japan) according to the manufacturer’s instructions. The principle of this method was reported previously [24].
Quantification of the SHAP-HA Complex and HA in BALF
The method was described previously [25]. Briefly, each well in a Maxisorp microtiter plate (Nunc, Roskilde, Denmark) was coated with 50 µl of 2 µg/ml HABPs (Seikagaku Corp., Tokyo, Japan) in 0.1 M sodium carbonate buffer (pH 9.40) at 4°C overnight, followed by blocking with 200 µl each of 2% BSA in phosphate-buffered saline with 0.1% (v/v) Tween 20 (PBS-T) at room temperature for 2 h. After washing with PBS-T, BALF samples (50 µl each) and standard SHAP-HA or HA solutions were applied to the wells, followed by incubation at 37°C for 1 h. After washing, 50 µl of rabbit anti-human IαI antibody (Dako, Glostrup, Denmark) (diluted 1:2,000 with PBS-T) or biotinylated-HABP (Seikagaku Corp., Tokyo, Japan) (0.5 µg/ml, diluted with 1% BSA/PBS-T) was added to each well, followed by incubation at 37°C for 1 h. Then, either horseradish peroxidase-conjugated goat anti-rabbit IgG immunoglobulins (Jackson ImmunoResearch Laboratories, West Grove, Pa., USA) (diluted 1:2,000 with PBS-T) or peroxidase-conjugated streptavidin (Jackson ImmunoResearch Laboratories) (diluted 1:2,000 with 1% BSA/PBS-T) was added (50 µl each), followed by incubation at 37°C for 1 h. Finally, color development was achieved by incubation with 3,3′,5,5′-tetramethyl benzidine substrate (KPL, Gaithersburg, Md., USA) (50 µl each) at 37 °C for 10 min and then stopped by adding 50 µl of 1 M HCl. The absorbance at 450 nm was measured on a VERSAmax microplate reader (Molecular Devices, Sunnyvale, Calif., USA). The assays were done in duplicate.
Analysis of Cytokine Levels in BALF
We measured TGF-β1 in BALF from each animal with an ELISA kit (eBioscience Inc., San Diego, Calif., USA) according to the manufacturer’s instructions. The other cytokines were measured with a cytokine antibody array (RayBiotech Inc.,Norcross, Ga., USA) according to the manufacturer’s instructions. The BALFs were pooled from mice in the same treatment groups, i.e. from 6 mice in the control group, from 7 mice in the WT group and from 7 mice in the KO group, and 1.5 ml of the pooled BALF in the respective groups were used for cytokine measurement.
The chemiluminescence was quantified with a LAS-4000 mini EPUV (Fujifilm, Japan) and Multi Gauge version 3.0 software (Fujifilm, Japan).
Histological Study of Lung Tissues
The left lung lobes were fixed in Histochoice Tissue Fixative (Sigma, Amresco Inc., USA) for 72 h and then embedded in paraffin. Serial 6-µm sections were subjected to hematoxylin and eosin (HE) staining, Giemsa staining, elastica-van Gieson (EVG) staining, periodic acid-Schiff (PAS) staining, histochemical staining for HA, and immunochemical staining for SHAP and CD44. The staining with biotinylated HABP, anti-SHAP antibodies, and anti-CD44 antibodies was done as described previously [20].
Immunostaining for sTNFR1, neutrophils and macrophages was also carried out, and the specimens were observed using a fluorescence microscope (Keyence BZ9000) (Keyence, Osaka, Japan). Macrophages were stained with anti-mouse macrophage monoclonal antibody (clone BM8) (BMA Biomedicals, Augst, Switzerland) and DyLight 549-conjugated goat anti-rat IgG (Rockland immunochemicals, Gilbertsville, Pa., USA). Neutrophils were stained with biotinylated anti-mouse Ly-6G rat IgG (clone RB6-8C5) (eBioscience). sTNFR1 was stained with rabbit anti-human sTNFR1 antibody (QED Bioscience Inc., San Diego, Calif., USA) which recognizes the soluble part of TNFR1, and Alexa Fluor 488-conjugated goat anti-rabbit IgG (H + L) (Molecular Probes).
Two blinded examiners evaluated randomly assigned slides.
Statistical Analysis
Results are presented as mean ± standard error. Significance was tested using the two-tailed Student’s t test or the Mann-Whitney U test; p < 0.05 was considered statistically significant.
Results
Increase in Airway Resistance in KO Mice
Airway response to MCh was measured by plethysmography (Penh) as described in Materials and Methods. In the control group, the Penh value at each concentration of MCh was lower on day 37 than on day 22 over all the concentrations of MCh. The reason for this phenomenon was not clear, but might be due to so-called acclimatization. This phenomenon would probably also occur in OVA-treated WT and KO mice. Therefore, the Penh values were compared among the animal groups at days 22 and 37.
Penh values were similar in KO and WT mice at day 22 (fig. 2a). After 2 weeks of OVA inhalation, airway hyperresponsiveness was successfully induced as manifested by the increased Penh value in both KO and WT mice (fig. 2b). At this time point, the Penh values of KO mice were significantly higher than those of WT mice, indicating that the KO mice had developed more severe airway hyperresponsiveness.

Airway responsiveness to inhaled MCh. a Airway responsiveness before OVA challenge. b Airway responsiveness after exposure to inhaled OVA for 2 weeks. * p < 0.05, ** p < 0.01.
Airway responsiveness to inhaled MCh. a Airway responsiveness before OVA challenge. b Airway responsiveness after exposure to inhaled OVA for 2 weeks. * p < 0.05, ** p < 0.01.
Enhancement of LPR but Not EPR to OVA in KO Mice
We investigated the specific airway responses to OVA, which are shown in figure 3. The EPR was comparable in KO and WT mice (fig. 3a). However, the LPR was significantly enhanced in KO mice (fig. 3b). We carried out another series of experiments using WT and KO mice both of which received saline plus aluminum hydroxide injections and were exposed to saline aerosol. The percent increase in Penh of EPR or LPR did not differ between the two animal groups.

EPR and LPR to OVA provocation. Saline-treated WT mice served as controls. a Following OVA inhalation for 30 min, no significant difference was found between EPRs of KO and WT mice (NS). b Following OVA challenge, the KO mice showed a significantly higher LPR than the WT mice. ** p < 0.01, *** p < 0.001.
EPR and LPR to OVA provocation. Saline-treated WT mice served as controls. a Following OVA inhalation for 30 min, no significant difference was found between EPRs of KO and WT mice (NS). b Following OVA challenge, the KO mice showed a significantly higher LPR than the WT mice. ** p < 0.01, *** p < 0.001.
Effect of SHAP-HA Deficiency on the Serum Levels of Allergen-Specific IgE and IgG1
To examine the underlining mechanisms for the difference of LPR to OVA between the KO and the WT groups, we measured OVA-specific IgE and IgG1, both of which were not detected in control mice treated with saline. IgE levels were increased to a similar level in OVA-treated WT and KO mice (756 ± 135 U/ml, n = 7 and 721 ± 211 U/ml, n = 7, respectively) (fig. 4a). IgG1 levels were also increased in both OVA-treated WT and KO mice, and were significantly higher in KO than in WT mice (2,120 ± 239 U/ml, n = 7 and 1,626 ± 103 U/ml, n = 7, respectively, p < 0.001) (fig. 4b).
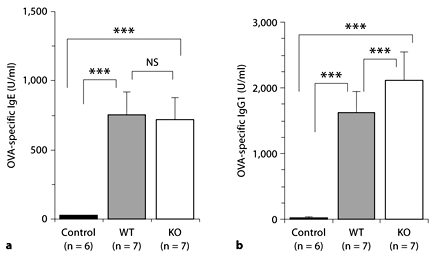
Measurement of OVA-specific serum IgE (a) and IgG1 (b). No significant difference was found in the serum levels of OVA-specific IgE between the KO and WT mice (NS). However, a higher (*** p < 0.001) serum level of OVA-specific IgG1 was found in the KO mice compared with WT mice. Saline-treated WT mice served as controls.
Measurement of OVA-specific serum IgE (a) and IgG1 (b). No significant difference was found in the serum levels of OVA-specific IgE between the KO and WT mice (NS). However, a higher (*** p < 0.001) serum level of OVA-specific IgG1 was found in the KO mice compared with WT mice. Saline-treated WT mice served as controls.
More Inflammatory Cells in BALF from KO Mice
Very few cells were found in the BALF from saline-treated control mice, most of which were macrophages and airway epithelial cells (data not shown). On the other hand, the total number of BALF inflammatory cells increased in the OVA-treated mice, indicating that OVA challenge induced a lung inflammation associated with inflammatory cell infiltration. However, the total number of inflammatory cells was not significantly different between in KO (761 ± 93/µl, n = 7) and in WT mice (641 ± 121/µl, n = 7) (fig. 5a). Differential cell counts showed that the numbers of eosinophils and lymphocytes were similar whereas the numbers of macrophages and neutrophils were significantly higher in KO mice (p < 0.05) (fig. 5b).

Total cell count (a) and differential cell count (b) in BALF. a The total cell number was not significantly different between KO and WT mice (NS). Very few cells were observed in the saline-treated control mice. b The numbers of alveolar macrophages (AM) and neutrophils (Neu) were significantly larger in KO mice than in WT mice (* p < 0.05, Mann-Whitney U test). No significant difference was found in the numbers of lymphocytes (Ly) and eosinophils (Eo).
Total cell count (a) and differential cell count (b) in BALF. a The total cell number was not significantly different between KO and WT mice (NS). Very few cells were observed in the saline-treated control mice. b The numbers of alveolar macrophages (AM) and neutrophils (Neu) were significantly larger in KO mice than in WT mice (* p < 0.05, Mann-Whitney U test). No significant difference was found in the numbers of lymphocytes (Ly) and eosinophils (Eo).
HA and SHAP-HA Levels in BALF
Sandwich ELISA demonstrated that small amounts of HA (4.8 ± 1.1 ng/ml) were present in the BALF of control mice. In contrast, OVA challenge induced dramatic (almost 10-fold) accumulations of HA (43.9 ± 30.7 ng/ml) in BALF in WT mice, but significantly less (19.8 ± 8.7 ng/ml) in KO mice (p < 0.05) (fig. 6a). OVA challenge also induced significant accumulation of the SHAP-HA complex in BALF in WT mice (p < 0.001) (fig. 6b). In BALF from KO mice, the SHAP-HA complex was not detected, confirming the deficiency in SHAP-HA complex formation in these mice.

Measurement of HA (a) and SHAP-HA (b) in BALF by sandwich ELISA. a The BALF of OVA-treated WT mice contained more HA than that of OVA-treated KO mice, and the BALF of both WT and KO mice contained more HA than that of saline-treated controls. b Larger amounts of SHAP-HA were found in the BALF from OVA-treated WT mice than from saline-treated WT mice. The BALF from KO mice did not contain any SHAP-HA. * p < 0.05, *** p < 0.001.
Measurement of HA (a) and SHAP-HA (b) in BALF by sandwich ELISA. a The BALF of OVA-treated WT mice contained more HA than that of OVA-treated KO mice, and the BALF of both WT and KO mice contained more HA than that of saline-treated controls. b Larger amounts of SHAP-HA were found in the BALF from OVA-treated WT mice than from saline-treated WT mice. The BALF from KO mice did not contain any SHAP-HA. * p < 0.05, *** p < 0.001.
Cytokine Levels in BALF
Cytokine levels in BALF were measured with a cytokine antibody array kit (fig. 7a) and a TGF-β1 ELISA kit (fig. 7b) as described in Materials and Methods. No apparent differences were found between the levels of IL-4, IL-5, IL-9, IL-10, IL-13, IFN-γ, and TNF-α in KO and WT mice. However, the OVA challenge induced an increase in the levels of IL-12p40 and sTNFR1 in both WT and KO mice; interestingly, this increase was higher in BALF from WT mice than in BALF from KO mice (compared to control mice: IL-12p40: 9-fold in WT mice and 3-fold in KO mice, and sTNFR1: 7-fold in WT mice and 4.6-fold in KO mice) (fig. 7a). The level of TGF-β1 also increased in the OVA-treated mice, but there was no significant difference between WT and KO mice (fig. 7b).

Determination of cytokines in BALF. a The cytokines in BALF were tested by a cytokine antibody array and analyzed by Multi Gauge version 3.0 for quantification. The BALFs were collected and pooled from mice in the same treatment groups, i.e. 6 animals in the control group, 7 animals in the WT group and 7 animals in the KO group. The levels of sTNFR-1 and IL-12p40 were lower in KO mice than in WT mice. b TGF-β1 concentration in BALF. No significant difference was found between KO and WT mice (NS), but both showed statistically significantly (p < 0.05) higher TGF-β1 concentrations than controls. Immunostaining was done twice and the determination was done in triplicate.
Determination of cytokines in BALF. a The cytokines in BALF were tested by a cytokine antibody array and analyzed by Multi Gauge version 3.0 for quantification. The BALFs were collected and pooled from mice in the same treatment groups, i.e. 6 animals in the control group, 7 animals in the WT group and 7 animals in the KO group. The levels of sTNFR-1 and IL-12p40 were lower in KO mice than in WT mice. b TGF-β1 concentration in BALF. No significant difference was found between KO and WT mice (NS), but both showed statistically significantly (p < 0.05) higher TGF-β1 concentrations than controls. Immunostaining was done twice and the determination was done in triplicate.
Histological Study and Immunostaining of Lung Tissues
Histological (HE, Giemsa, PAS and EVG staining) and immunochemical analyses (HA, CD44 and SHAP-HA staining) were performed on lung tissues to examine the inflammation and airway remodeling. Histological staining showed that more inflammatory cells around airways, more goblet cell hyperplasia in airway epithelia, and more thickened basement membranes were present in OVA-treated lungs than in saline-treated control lungs (fig. 8a). In order to explain the elevated number of neutrophils and macrophages in BALF from the KO mice, we carried out immunostaining for neutrophils and macrophages in the lung tissues. However, the amount of infiltrating neutrophils or macrophages did not seem different between the KO and the WT mice (data not shown).

Histological (a) and immunochemical (b) staining of lung specimens of OVA-treated WT and KO mice and of saline-treated WT control mice. a Representative sections stained with HE, PAS, Giemsa and EVG. The patterns suggest airway inflammation, goblet cell hyperplasia and airway fibrosis in OVA-treated lungs. Magnification: ×20 for all photographs. b Representative staining for HA, CD44, SHAP-HA complex and sTNFR1. Strong HA staining may be observed in the lungs of OVA-treated mice compared with the lung of a saline-treated WT mouse. In the staining for CD44, the lungs of OVA-treated WT and KO mice show similar staining levels; the lung of a saline-treated WT control shows weaker staining. Obvious SHAP-HA complex staining may be observed around the airways of an OVA-treated WT mouse, but there is no significant staining around those of a saline-treated WT mouse. There is no staining in the KO mouse lung. In the immunostaining for sTNFR1, the specimens were observed using a fluorescence microscope. sTNFR1 appears to be upregulated in an OVA-treated mouse, and the stain is stronger in the specimen from a KO mouse than in that from a WT mouse. Magnification: ×20 for all photographs.
Histological (a) and immunochemical (b) staining of lung specimens of OVA-treated WT and KO mice and of saline-treated WT control mice. a Representative sections stained with HE, PAS, Giemsa and EVG. The patterns suggest airway inflammation, goblet cell hyperplasia and airway fibrosis in OVA-treated lungs. Magnification: ×20 for all photographs. b Representative staining for HA, CD44, SHAP-HA complex and sTNFR1. Strong HA staining may be observed in the lungs of OVA-treated mice compared with the lung of a saline-treated WT mouse. In the staining for CD44, the lungs of OVA-treated WT and KO mice show similar staining levels; the lung of a saline-treated WT control shows weaker staining. Obvious SHAP-HA complex staining may be observed around the airways of an OVA-treated WT mouse, but there is no significant staining around those of a saline-treated WT mouse. There is no staining in the KO mouse lung. In the immunostaining for sTNFR1, the specimens were observed using a fluorescence microscope. sTNFR1 appears to be upregulated in an OVA-treated mouse, and the stain is stronger in the specimen from a KO mouse than in that from a WT mouse. Magnification: ×20 for all photographs.
HA and CD44 staining revealed upregulation of these molecules in OVA-treated lungs. However, no significant differences were found between the KO and WT mice (fig. 8b). Immunostaining of the SHAP-HA complex showed no significant signal in the sections from OVA-treated KO mice and saline-treated control mice. Obvious staining of SHAP-HA complex was found in the sections from OVA-treated WT mice (fig. 8b).
The sTNFR1 level in BALF from the KO mice was found to be lower than in BALF from the WT mice (fig. 7a). To examine the status of expression of this molecule in the lung tissues, immunochemical staining of sTNFR1 was carried out. As shown in figure 8b, the staining revealed upregulation of sTNFR1 in OVA-treated animals, but appeared to be stronger in the KO than in the WT mice.
Discussion
In the present study, Penh was used as an indicator of airway responsiveness. The advantage of this technique includes noninvasiveness and easy performance, the ability to obtain data rapidly and repeatedly, and the good correlation with lung resistance [22,23,26]. On the other hand, it has been objected in some reports that unrestrained plethysmography was an unreliable measurement technique and that Penh, dominated by conditioning, was essentially unrelated to lung resistance in some mouse strains [27,28]. However, Lomask [23 ]argued that flow plethysmography, in contrast to pressure plethysmography, was a stable and reliable measurement of airway responsiveness. Indeed, critical studies on the correlation of airway resistance to Penh revealed a satisfying correlation in BALB/c mice [28]. In the present study, where BALB/c mice were used, Penh, measured by flow plethysmography, stably increased with increasing concentrations of MCh in both placebo- and OVA-treated groups. Therefore, we consider that in our study Penh was a reliable parameter for evaluating airflow limitation and airway responsiveness.
We induced murine asthma by OVA sensitization and challenge. It was reported that OVA-sensitized mice are good models for airway hyperresponsiveness, but not for acute response to inhaled allergen after OVA challenge because of limited EPR and LPR in BALB/c mice [29]. In our study, airway hyperresponsiveness to MCh was successfully induced in both KO and WT mice, with the extent of increase significantly larger in KO mice than in WT mice. In the acute response to inhaled OVA, neither EPR nor LPR was observed in WT mice. The KO mice showed no EPR, but an LPR was clearly detected. These findings correlate well with the asthma model reported by Zosky et al. [29]. Sera from KO mice showed a similar level of IgE and a higher level of IgG1 compared to WT mice. It has been reported that in mice immunized with coagulated egg white, the OVA-induced cutaneous LPR in the paws was associated with high serum levels of OVA-specific IgG1, but not IgE [30]. Although the mechanisms of LPR are not clearly understood, LPR might rather be correlated with IgG1-mediated immediate allergic response under certain circumstances, such as in the SHAP-HA-deficient condition.
Inflammatory cell infiltration is also one of the important aspects in allergic asthma. In the present study, we observed that the number of neutrophils in BALF from the KO mice was higher than that from the WT mice. However, the immunostaining of neutrophils in the lung tissues revealed no detectable difference between the two groups. SHAP has been reported to potentiate the CD44-mediated leukocyte adhesion in the synovium [20]. Thus, the elevated number of neutrophils in BALF from the KO mice might be tentatively explained by the fact that neutrophils easily emigrate to BALF in the absence of SHAP.
In the present study, histological studies demonstrated comparable airway remodeling in KO and WT mice. This is consistent with a previous study reporting that the extent of fibrosis in bleomycin-induced fibrotic lung injury was comparable between KO and WT mice [21]. However, the possibility remains that a 2-week challenge of allergen is too short to induce tissue remodeling extensive enough to manifest the effect of the SHAP-HA complex. The supporting results are that KO mice showed higher airway hyperresponsiveness to MCh challenge and a higher LPR to OVA provocation.
HA fragmentation is often associated with inflammatory responses, including pulmonary inflammation [14]. Low-molecular-weight HA has a proinflammatory role by evoking eosinophils and preventing apoptosis of inflammatory cells [31,32,33]. The observation that the HA levels in BALF are lower in KO mice than in WT mice seems opposite to the other results showing enhanced inflammation in KO mice. It is noteworthy that HA was quantified with a sandwich ELISA, which does not detect small HA fragments of thousand kilodaltons very well, which may have been present. Indeed, HA staining in lung sections was comparable between KO and WT mice, suggesting that the upregulation of HA production upon inflammation was not significantly altered in KO mice. In addition, it has been suggested that formation of the SHAP-HA complex may prevent the fragmentation of HA [34]. In this respect, the enhanced inflammation in KO mice may be partially due to increased HA fragmentation in the absence of SHAP.
The present study also revealed lower IL-12p40 levels in the BALF from KO mice. The homodimer of IL-12p40 has been reported to bind to the common receptor IL-12Rβ1 [35,36], and to inhibit IL-12-mediated functions, which could serve as a negative feedback loop in vivo [37,38]. Thus, low levels of IL-12p40 in KO mice may attenuate its inhibitory effect on IL-12 functions. IL-12 is generally known to promote differentiation to Th1 lymphocytes and eventually to counter-regulate the effect of Th2 cells, which have critical roles in the progression of allergic inflammations. In addition, there is also evidence that Th1 cell responses (e.g. secretion of TNF-α and IFN-γ) might also be responsible for some pathogenic features of chronic allergy [39]. Therefore, it is likely that the low IL-12p40 levels underlie the increased airway hyperresponsiveness in KO mice. However, since no apparent difference was found in the BALF levels of Th2 cytokines (i.e. IL-4, IL-5, IL-9, IL-10 and IL-13) and Th1 cytokines (i.e. TNF-α and IFN-γ) examined in this study, the mechanism remains to be investigated further. Clarifying the status of cytokine productions for explanation of the cytokine levels in BALF is very important and needs to be investigated.
TNF-α has been reported to promote airway inflammation, induce airway hyperresponsiveness, and to have a central role in airway remodeling [40,41,42]. Consistently, TNF-α blockade effectively inhibits airway hyperresponsiveness and airway inflammation [43,44], and in a series of clinical trials asthma was improved by anti-TNF-α therapy [45,46]. sTNFR1, recognized as a TNF-α blockade, binds to TNF-α and competitively inhibits its function [47,48]. In the present study, similar levels of TNF-α, but lower levels of sTNFR1, were found in the BALF from KO mice, indicating that the net TNF-α activity is likely to be significantly higher in KO mice. Recently, TNF-α was shown to increase CD38 expression and protein activity in airway smooth muscle cells, which increased responses to a variety of contractile agonists due to alteration in homeostasis of intracellular calcium ion [49,50]. Accordingly, higher TNF-α activity in the absence of the SHAP-HA complex might contribute to the high contractility of airway smooth muscle as well as to allergic inflammation with resulting increased airway hyperresponsiveness to MCh in the present study. Further, a decrease in the stability of the extracellular matrix in SHAP-HA deficiency might also result in a decrease in stiffness of airway walls, and thereby increase airflow limitations by MCh, although further studies are needed.
In the immunochemical study of the lung tissues, we also found that the staining of sTNFR1 was stronger in the KO than WT mice. As to the lower sTNFR1 levels in BALF of the KO than in the WT mice, it may be possible that the upregulation of sTNFR1 is the same in KO and the WT mice; however, a larger amount of sTNFR1 may be secreted into the BALF in WT mice than in KO mice. In other words, shedding of sTNFR1 from the cell surface may be promoted in the presence of the SHAP-HA complex in this murine model of OVA-induced asthma.
The KO mice used in this study are deficient in the SHAP-HA complex as well as in bikunin. Since bikunin shows a weak inhibitory activity against a broad spectrum of proteases, the protease/inhibitor balance may tilt in the absence of bikunin, theoretically resulting in a weak nonspecific strengthening of inflammation. However, we think such a mechanism is unlikely to have played a role in the present study because the inhibitory activity of bikunin is generally very weak, accounting for no more than 5% of the total plasma inhibitory activity, and free bikunin is rapidly cleared from the circulation [51,52].
In summary, we identified an essential role for the SHAP-HA complex in a murine model of OVA-induced asthma. The SHAP-HA complex can stabilize airway extracellular matrix and promote the secretion of sTNFR1, a TNF-α blockade in allergic asthma, thus exerting an inhibitory function on development of excessive airway hyperresponsiveness and probably on airway allergic inflammation. Further studies will focus on how the absence of the SHAP-HA complex results in the decrease in sTNFR1 and IL-12.
Acknowledgements
This study is supported in part by a Grant-in-Aid for Scientific Research (B and C) from the Japan Society for the Promotion of Science (B No. 17370041 for K.K., C No. 17590807 to E.Y. and C No. 21570147 for L. Zhuo), a grant from Akeikai Foundation (L. Zhuo), and also by NIH AI067816 (M.A.A.) and HL081064 (V.C.H. and M.A.A.).
