

Abstract
Alcohol abuse is a common cause of both acute and chronic pancreatitis. There is a wide spectrum of pancreatic manifestations in heavy drinkers from no apparent disease in most individuals to acute inflammatory and necrotizing pancreatitis in a minority of individuals with some progressing to chronic pancreatitis characterized by replacement of the gland by fibrosis and chronic inflammation. Both smoking and African-American ethnicity are associated with increased risk of alcoholic pancreatitis. In this review we describe how our recent studies demonstrate that ethanol feeding in rodents causes oxidative stress in the endoplasmic reticulum (ER) of the digestive enzyme synthesizing acinar cell of the exocrine pancreas. This ER stress is attenuated by a robust unfolded protein response (UPR) involving X-box binding protein-1 (XBP1) in the acinar cell. When the UPR activation is prevented by genetic reduction in XBP1, ethanol feeding causes significant pathological responses in the pancreas. These results suggest that the reason most individuals who drink alcohol heavily do not get significant pancreatic disease is because the pancreas mounts an adaptive UPR to attenuate the ER stress that ethanol causes. We hypothesize that disease in the pancreas results when the UPR is insufficiently robust to alleviate the ER stress caused by alcohol abuse.
Epidemiology of Alcoholic Pancreatitis: Emerging Role for Smoking
Alcohol abuse is associated with both acute and chronic pancreatitis, with alcohol abuse being the most common cause of chronic pancreatitis in European countries, the USA, Japan, Korea and Australia [1,2,3,4,5,6,7,8,9,10,11,12]. In contrast, idiopathic pancreatitis is the most common stated etiology in India and China [9,13]. Of note, in countries where alcohol abuse-induced chronic pancreatitis is more prevalent than idiopathic pancreatitis the prevalence of pancreatitis due to alcohol abuse is predominantly seen in men, with idiopathic pancreatitis more commonly reported in women [9,10].
Of particular importance, when considering diseases of organ injury, pancreatitis is the most common alcohol-related hospital diagnosis in the USA [14]. The incidence and prevalence are difficult to ascertain with precision because of the variable nature of the disease presentation including the fact that alcohol abuse causes a continuum of disease manifestations from acute to chronic pancreatitis. Importantly, findings consistent with pancreatitis have been demonstrated in up to 75% of autopsies done on alcoholics although clinical pancreatitis is diagnosed in less than 10% of alcoholic patients [4,15]. This finding suggests that pancreatitis occurs without clinical manifestations in a large proportion of heavy drinkers.
The risk of developing alcohol-induced pancreatitis is related to the amount and duration of drinking. A minimum of 6–12 years of drinking 80 g or greater amounts per day of alcohol are generally considered required for development of the disease [4,5,16]. There are ethnic, dietary and smoking co-factors that contribute to the risk of developing pancreatic disease from alcohol abuse. For example, recent reports show that African-American patients are 2–3 times more likely than Hispanic, Caucasian and Asian patients to be hospitalized for alcohol-related pancreatitis than alcoholic cirrhosis [14,17].
Smoking is emerging as a major risk factor for alcoholic pancreatitis. Although the complicated interrelationships between smoking and drinking make this area of epidemiology challenging [4], careful epidemiologic studies [16,18,19,20,21] are demonstrating that cigarette smoking is an independent risk factor for alcohol abuse-associated pancreatitis, that smoking may have a synergistic effect with alcohol abuse to cause pancreatitis, and that smoking accelerates the progression of the disease.
Other factors such as diet may contribute to the modulation and progression of alcoholic pancreatitis. For example, some reports indicate that diets high in protein and fat are positively associated with the disease [22,23] whereas increased vitamin E and coffee intake are negatively associated with the disease [18,24].
In sum, there is wide variability in the reporting of the incidence and prevalence of alcohol abuse-induced pancreatitis so that determining risk factors and their relative contributions to disease development are difficult to ascertain with precision. However, there are now established risk factors related to ethnicity and smoking. As described below, the mechanisms involved in the pathobiology of alcohol-induced pancreatic diseases are incompletely understood. Importantly, the roles ethnicity and smoking play in the mechanisms of disease are not understood at all. However, incorporating these co-factors into our models designed to reveal mechanism will likely provide further insights into the pathobiology of alcoholic pancreatitis.
Pathobiology of Alcoholic Pancreatitis
As indicated above, alcohol abuse is associated with a full spectrum of pancreatic responses. In the majority of alcohol abusers there are no clinical manifestations of pancreatic disease. In those who develop pancreatic disease findings there are findings of both acute and chronic pancreatitis [1,2]. In general, early after the onset of pancreatic disease the pancreas shows inflammation and necrosis of tissue and a minimum of chronic inflammation and fibrosis. In contrast, specimens of pancreas several years after the onset of pancreatic disease from alcohol abuse show severe fibrosis and chronic inflammation replacing normal exocrine pancreatic tissue [25,26,27].
Although the initial mechanisms leading to these pathologic responses to alcohol abuse are incompletely understood, several effects of alcohol and its metabolites have been found in experimental systems to explain some of the pathology due to alcohol abuse. These findings include the fact that alcohol promotes inappropriate intracellular activation of digestive enzymes [28,29] and misdirection of digestive enzyme secretion to the basolateral surface of the acinar cell [30,31,32,33,34], and that ethanol and one of its key metabolites, fatty acid ethanol ester, promote intracellular activation of digestive enzymes, acute inflammation and necrosis of pancreatic cells and tissues [35,36,37,38,39,40,41,42,43,44,45]. Ethanol and fatty acid ethanol ester can cause fibrosis through activation of stellate cells and preventing degradation of the extracellular matrix proteins that these activated cells produce [46,47,48,49,50,51,52]. Finally, alcohol alters the immune response preventing resolution of the acute inflammatory response and promoting a chronic inflammatory response [53,54,55].
Below we discuss our early findings related to the effects of ethanol feeding on endoplasmic reticulum (ER) stress and the unfolded protein response (UPR). These findings are provocative and may explain some of the early events in alcohol abuse-mediated injury. We hypothesize that disease in the pancreas results when the UPR is insufficiently robust to alleviate the ER stress caused by alcohol abuse.
Endoplasmic Reticulum Stress and the Unfolded Protein Response
The acinar cell of the exocrine pancreas has a highly developed ER system for the synthesis and secretion of digestive enzymes [56,57]. Each protein synthesized in the ER must undergo a specific folding sequence involving biochemical modifications requiring chaperones and foldases before it can be transported to destination organelles such as Golgi, zymogen granule (digestive enzyme storage granules) and lysosome, or membrane sites. These processes require optimal redox conditions and ion concentrations (in particular calcium) for the enzymes involved to accomplish their biochemical reactions. These processes are likely impacted by variations in demand for protein synthesis and environmental factors impacting the ER including alcohol, smoking, and xenobiotics. How the ER addresses these issues falls under the topics of ER stress and the UPR.
The ER of the acinar cell in the exocrine pancreas requires a robust UPR system considering the fact that its protein synthesis demands are the greatest of any tissue in the body. There is information from previous studies [58] showing the presence and activation of the UPR during experimental pancreatitis. However, ER stress and UPR have not been studied in the exocrine pancreas in response to the environmental factors that predispose to pancreatitis listed above.
In general, ER stressors include genetic mutation of cellular proteins, reactive oxygen species (ROS), metabolic modifications of nascent proteins in the ER and/or alterations in the concentrations of ions in the luminal ER (i.e. calcium) [59]. These perturbations lead to unfolding/misfolding nascent proteins in the ER lumen. There are three major adaptive UPR systems that sense the unfolded/misfolded proteins via ER transmembrane proteins and transmit this information to signaling systems to address the ER stress. These have been reviewed extensively elsewhere [59,60,61,62,63,64]. For the purposes of this paper, the three systems will be only briefly described. Of note, the mechanism of sensing perturbations in ER proteins and the ER luminal environment are not well established.
The overall functions of these three systems are to: (1) upregulate the expression and function of foldases and chaperones needed to augment the folding and export capacity of newly synthesized proteins of the ER; (2) activate a protein degradation system called ER-associated protein degradation (ERAD) to rid the ER of accumulated unfolded and misfolded proteins; and (3) reduce translation of mRNA to decrease the processing demand for newly synthesized proteins.
One system involves the ER transmembrane sensor, inositol-requiring protein-1 (IRE-1). ER stress induces homodimerization of IRE-1 which activates its RNAse activity. The RNase activity is specifically targeted to the mRNA for unspliced X-box binding protein-1 (XBP1) removing a 26 nucleotide intron resulting in an mRNA that translates into a potent transcription factor, spliced XBP1 (XBP1-S). XBP1-S transcriptional regulation leads to increased expression of chaperones such as the binding immunoglobulin protein (BiP) and glucose regulated protein-94 (GRP94); foldases such as protein disulfide isomerase (PDI); ERAD components, and XBP1. XBP1-S also upregulates synthesis of membrane lipids needed for ER expansion needed to adapt to ER stress.
A second system utilizes the ER transmembrane sensor, activating transcription factor-6 (ATF6). This C-terminal luminal domain of ATF6 is responsive to ER stress resulting in its release from the ER membrane and transport to the Golgi compartment where it is cleaved by proteases. The cleavage provides an N-terminal fragment that is a transcription factor that increases the expression of XBP1 as well as other UPR target genes. Thus, ATF6 acts in concert with IRE-1 to elicit a XBP1-mediated UPR.
The third system of the UPR is protein kinase-like endoplasmic reticulum kinase (PERK). This sensor responds to ER stress by causing a significant translational inhibition of protein synthesis. PERK activation involves its autophosphorylation resulting in kinase activity directed to the α-subunit of eukaryotic translation initiation factor-2nα (eIF-2α). In its activated and phosphorylated form eIF-2α is essential for translation because it initiates translation of each mRNA through recruitment of the first tRNA (tRNAMET) to ribosomal subunits to start translation. An additional effect of persistent activated phosphorylated eIF-2α is the specific translational upregulation of activating transcription factor-4 (ATF4) that targets genes promoting antioxidant effects such as the synthesis of glutathione, and expression of the transcription factor C/EBP homologous protein (CHOP) which induces apoptotic cell death.
As illustrated in the description below, each of these three ER transmembrane sensors are affected in our animal models designed to determine the effects of alcohol abuse on the exocrine pancreas.
Alcohol, ER Stress and the UPR
Long-term feeding of ethanol alone causes minimal pancreatic tissue injury in animal models [55,65]. To account for this lack of injurious effect, we hypothesized that ethanol feeding causes ER stress and that a physiologic adaptive UPR responds to the ER stress preventing pathobiologic pancreatitis responses such as inflammation and cell death. As indicated above, previous studies [58] demonstrated that all three ER stress/UPR transducers (i.e. IRE-1, ATF6 and PERK) and their downstream pathways are activated in experimental pancreatitis. However, there was no information on the nature of the ER stressors activating the UPR responses in the models of experimental pancreatitis. More importantly, there was no information on how the pancreas responds physiologically to ER stressors generated by alcohol abuse or other potentially toxic factors such as smoking.
To test our hypothesis, we designed a series of studies to characterize the effect of ethanol feeding on development of ER stress and the UPR, and determine the importance of the UPR in physiologic adaptation and prevention of pathologic consequences [66]. We used rats and mice fed control diets or ethanol-containing diets for 4–6 weeks using the Tsukamoto-French intragastric model [67] which provides continuous feeding of the diets to the animals. With this feeding protocol we found no obvious pancreatic damage using light microscopic examination of the tissue. However, a careful examination by electron microscopy demonstrated extensive dilation of the ER of acinar cells, an indicator of ER stress. In addition, measurements of the redox status of the ER found that there was decreased reduced glutathione and increased oxidized glutathione in the ethanol-fed rats indicating that the alcohol feeding generates ROS in the ER of the pancreatic acinar cell. Thus, ROS represent at least one set of ER stressors.
In an evaluation of the sensors and signals of the UPR, we found that ethanol feeding increased the expression of IRE-1 and XBP1-S along with a small increase in PERK activation [66]. In addition, alcohol feeding significantly upregulated a key oxidoreductase, PDI. Finally, ethanol feeding altered PDI structure so that a greater proportion of the PDI was in its oxidized state in the ethanol-fed animals compared to the control-fed.
PDI is an oxoreductase that is critical for protein folding by catalyzing disulfide bond formation, a key step for maturation of proteins in the secretory pathway [68]. This function is often referred to as ‘oxidative folding’ because PDI catalytic action requires reduced glutathione to sustain its ability to regenerate and sustain its ability to repeatedly form disulfide bridges [69].
The combination of findings listed above indicates that ethanol feeding leads to an oxidative state in the ER of the acinar cell. The oxidative state could come from one or more biochemical mechanisms. For example, oxidative ethanol metabolism in the ER could directly lead to the oxidative states. On the other hand, ethanol and/or its metabolites could lead to modifications of nascent proteins in the ER that frustrate oxidative folding so that PDI is ineffective in catalytic redox cycles leading to more utilization and depletion of reduced glutathione. The end result in either situation would be a net generation of ROS in the ER, decreased reduced glutathione and increased oxidized glutathione and PDI itself.
Considering the results described above, we hypothesized that the effect of alcohol feeding to increase expression of IRE-1 and XBP1-S is necessary for the acinar cell to adapt to ER stress resulting from alcohol abuse. We tested this hypothesis using animals with heterozygous deficiency for XBP1. We chose this approach because homozygous depletion of XBP1 results in lethality, and because we hoped for a situation where we could specifically prevent the increase in XBP1 that we observed with ethanol feeding.
In the heterozygous animals, the ethanol feeding led to levels of XBP1-S expression similar to that observed in wild-type animals receiving the control diet allowing us to specifically observe the response of the pancreatic tissue in the absence of the UPR to ethanol feeding. In contrast to the effect in wild-type animals, ethanol feeding resulted in marked morphological and biochemical effects in animals with heterozygous deletion of XBP1.
In the pancreatic tissue of the XBP1-deficient animals receiving alcohol there was a seriously disorganized ultrastructure with a decreased number of zymogen granules, and the remaining ones were inappropriately scattered throughout the cell and not localized to their normal apical position. There was extensive dilation of the ER with occasional dense luminal inclusions, hallmarks of ER stress, as well as significant accumulation of autophagic vacuoles. By electron microscope examination, these abnormalities could be found in more than 40% of total acinar cells. There was also a marked decrease in levels of the digestive enzyme amylase corresponding to the decreased number of digestive enzyme-storing zymogen granules. Finally, about 20% of the pancreas contained lesions representing severe injury. These areas showed acinar cell necrosis, apoptosis and inflammation with replacement by stromal cells and ductular-appearing regenerating cellular structures.
In ethanol-fed animals the XBP1 deficiency prevented the increase in PDI while markedly enhancing PERK and eIF2α phosphorylation and expression of ATF4, all features of prolonged, severe and unchecked ER stress [69]. These signals mediate translational inhibition (PERK and eIF2α phosphorylation) and expression of CHOP (ATF4) accounting for the decrease in digestive enzyme and increased cell death that we observed. Ethanol feeding in the XBP1-deficient animals also led to a decrease in expression of EDEM1, a key participant in the ERAD pathway for degradation for unfolded and misfolded proteins. Such a decrease in ERAD may account for the marked increase in autophagy in the pancreas of these animals because previous studies [70] have shown that EDAM1 deficiency enhances autophagy to dispose of misfolded proteins.
Conclusions
The results of the studies in ethanol-fed animals reviewed here provide insights into how the exocrine pancreas adapts its secretory machinery in the face of alcohol abuse so that pathologic consequences are infrequent. As summarized in figure 1, alcohol abuse results in an oxidative environment in the ER either directly from the metabolism of ethanol or through generation of ROS because of ethanol-mediated alterations in nascent proteins causing futile catalytic redox cycles. By mechanisms that are not known yet, the oxidative environment in the ER leads to increased expression of IRE-1 and splicing of XBP1 for translation into the active transcription factor, XBP1-S. This transcription factor, in turn, mediates the expression of chaperones, oxidoreductases such as PDI, ERAD proteins and increased lipid synthesis to expand the capacity of the ER. This UPR leads to attenuation of the ER stress.
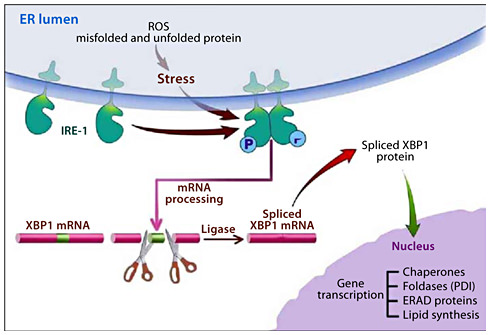
ER stress with alcohol abuse and the adaptive UPR. Alcohol metabolism causes increased ROS leading to unfolded and misfolded nascent proteins in the ER. The ER stress also results in activation of an adaptive UPR led by activation of IRE-1 through homodimerization. Activated IRE-1 is an RNAse directed to splicing out in intron of XBP1 leading to XBP1-S mRNA. XBP1-S is translated into a potent transcription factor that regulates the expression of chaperones, foldases including PDI, components of the ERAD system and enzymes of lipid synthesis needed to expand the ER. These effects adapt the ER to the ER stress caused by alcohol abuse.
ER stress with alcohol abuse and the adaptive UPR. Alcohol metabolism causes increased ROS leading to unfolded and misfolded nascent proteins in the ER. The ER stress also results in activation of an adaptive UPR led by activation of IRE-1 through homodimerization. Activated IRE-1 is an RNAse directed to splicing out in intron of XBP1 leading to XBP1-S mRNA. XBP1-S is translated into a potent transcription factor that regulates the expression of chaperones, foldases including PDI, components of the ERAD system and enzymes of lipid synthesis needed to expand the ER. These effects adapt the ER to the ER stress caused by alcohol abuse.
When the UPR is attenuated by preventing the increase in XBP1-S through genetic deletion, there is unresolved and augmented ER stress resulting in PERK activation causing global protein synthesis inhibition and ATF4-mediated activation of cell death pathways via CHOP expression. The inhibition of the UPR also results in marked cellular and tissue pathology that represents aspects of alcoholic pancreatitis.
These results may provide an explanation at least in part why only a small percentage of heavy drinkers develop pancreatitis. We speculate that those that develop disease have an insufficient UPR to resolve the oxidative ER stress caused by alcohol abuse. This could occur as a result of co-factors such as smoking and/or dietary factors that could increase ER stress above the capacity of a normal UPR to resolve, genetic polymorphisms in one or more components of the UPR making the system less capable of responding to the ER stress, and/or environmentally-induced alterations in the UPR preventing a sufficiently robust UPR. An example of genetic polymorphisms in a UPR component that alters disease risk was recently reported in inflammatory bowel disease [60,71]. In this case, there was a fourfold increase in relatively rare genetic polymorphisms in patients with inflammatory bowel disease compared to controls.
These results also allow us to speculate that other causes of pancreatitis including genetic mutations in digestive enzymes, smoking, drugs and metabolic disorders such as diabetes and hypertriglyceridemia may cause pancreatic disease by causing ER stress that is not sufficiently resolved by the UPR.
Acknowledgments
The authors acknowledge support from the Department of Veterans Affairs, Greater Los Angeles Healthcare System, USC-UCLA Southern California Research Center for ALPD and Cirrhosis (P60 AA11999), UCLA Center for Excellence in Pancreatic Diseases (P01AT003960), and National Institute of Health, NIDDK (1R21 AA016010-01).
Disclosure Statement
No financial or other conflicts of interest exist in relation to the content of the article.
