

Abstract
In a 24-year-old man with mild intellectual disability, congenital heart defects and obesity, we identified up to 4 small supernumerary marker chromosomes (sSMCs) in blood metaphases. The ring-shaped sSMCs were derived from chromosomes 11, 12 and X as well as a fourth, unidentified chromosome. In interphase nuclei of epithelial cells from the urinary tract and buccal mucosa, the presence of the r(11), r(12) and r(X) was confirmed by FISH. Using Illumina Infinium 317K SNP-arrays, we detected 3 copies of the pericentromeric regions of chromosomes 11, 12 and X. The r(X) was present in 84–89% of cells in the various tissues examined, lacks the XIST gene, but contains FAM123B, a potential dosage-sensitive candidate gene for congenital cardiac abnormalities, and ARHGEF9, a candidate gene for intellectual disability. ARHGEF9 encodes collybistin (CB), which is required for localization of the inhibitory receptor-anchoring protein gephyrin and for formation and maintenance of postsynaptic GABAA and glycine receptors. We propose that the 2-fold increase in dosage of ARHGEF9 disturbs the stoichiometry of CB with its interacting proteins at inhibitory postsynapses. SNP alleles and short tandem repeat markers on the r(11) and r(X) were compatible with a maternal origin of both sSMCs through a meiosis II error. The sSMCs may have resulted from predivision chromatid nondisjunction, leading to anaphase lagging, followed by incomplete degradation of the supernumerary chromosomes.
Small supernumerary marker chromosomes (sSMCs) are defined as additional centric derivatives smaller than chromosome 20 [Liehr et al., 2004]. Single sSMCs are detected in about 0.043% of unselected newborns, 0.077–0.11% of prenatal cases and in 0.29–0.43% of patients with intellectual disability [Liehr et al., 2004, 2006; Bartsch et al., 2005; Huang et al., 2006; Liehr and Weise, 2007; Forabosco et al., 2009]. Multiple sSMCs are among the rarest cytogenetic abnormalities. As estimated from reviewing 1,396 cases, roughly 1.4% of the cases with supernumerary markers have multiple sSMCs [Liehr et al., 2004], and, therefore, the frequency of such cases in patients with developmental delay can be estimated at 1 in 17,000–25,000. More than 30 cases have been described, as reviewed by Beverstock et al. [2003], Daniel and Malafiej [2003], Reddy et al. [2003], and Liehr et al. [2004]. Most of these cases concerned patients with developmental delay or intellectual disability, often in combination with dysmorphic signs and multiple congenital anomalies.
While several models for the generation of single sSMCs have been proposed [Liehr et al., 2004], the mechanism of origin of the multiple sSMCs is unknown. In 1 patient, these sSMCs are either small ring chromosomes, minute linear chromosomes, or both. In general, they contain centromeric heterochromatin and variable, but limited, amounts of pericentromeric euchromatin, either from the short arm, long arm, or both arms [Baldwin et al., 2008]. More recently, also sSMCs of a more complex architecture have been reported, containing material from 2 distinct chromosomes or non-contiguous regions from the same chromosome [Davidsson et al., 2010; Fei et al., 2011; Vetro et al., 2012]. The paucity of informative polymorphic DNA markers, together with chromosomal mosaicism, hampered the identification of the parental origin in cases with multiple sSMCs. As is evident from the literature [Beverstock et al., 2003; Daniel and Malafiej, 2003; Reddy et al., 2003; Liehr et al., 2004], any human chromosome can give rise to 1 of the multiple sSMCs in the patients described so far, with each patient appearing to harbor a unique set of sSMCs. Therefore, the origin of multiple sSMCs in an individual is most likely explained as the result of a single event, such as the breakdown and rearrangement of a haploid chromosome complement shortly after fertilization in a triploid zygote [Beverstock et al., 2003; Daniel and Malafiej, 2003].
Here, we report a new case of a patient with mild intellectual disability who has up to 4 small supernumerary ring chromosomes in metaphases from stimulated peripheral blood lymphocytes. Using FISH, array-CGH and array-based SNP-analysis, we determined the chromosomal origin and gene content of the sSMCs. In addition, using short tandem repeat (STR) and SNP markers, we investigated the parental origin of the multiple sSMCs.
Case Report
The patient is a 24-year-old male with mild developmental delay, who, from the age of 7 on, gradually became obese. At the age of 20, his body mass index was 33.1. He had a congenital secundum-type atrial septal defect, a hemodynamically significant perimembranous ventricular septal defect and dysplastic aortic and pulmonary valves. The septal defects were surgically corrected at the age of 11 and the redundant tissue of the pulmonary valves was excised as well. He was operated for bilateral inguinal hernias at age 1, underwent left-sided orchidopexy at the age of 14, and surgical correction for a left-sided club foot. A detailed case report has been published [Joziasse et al., 2009].
Materials and Methods
Molecular Cytogenetic Investigations
Slides containing metaphases from phytohemagglutinin-stimulated lymphocytes from peripheral blood were prepared following standard methods. Metaphases were analyzed with GTG-banding at the 550-band level. FISH was performed according to standard methods [Liehr and Claussen, 2002] except that, after hybridization, slides were washed twice in 0.4× SSC/0.05% Tween-20 at 72°C for 5 min, followed by washes in 2× SSC/0.05% Tween-20 and 4× SSC/0.05% Tween-20 at room temperature for 5 min each. FISH on epithelial cells from buccal smears and urine was performed according to Rauch [2002]. Centromere-specific DNA probes were obtained from Abbott-Vysis and hybridized according to instructions provided by the manufacturer. A FITC-labeled telomere-specific TTAGGG peptide nucleic acid probe was obtained from DAKO and used according to protocols provided by the manufacturer. Array-CGH was performed according to Poot et al. [2007a], using the BAC-array described by Vissers et al. [2003]. SNP analysis was performed as described earlier [Poot et al., 2007b] using the Infinium HumanHap317K Genotyping BeadChip (Illumina Inc.) [Poot et al., 2010]. Data about the gene content of the sSMCs were compiled using the Database of Genomic Variants (DGV: http://projects.tcag.ca/variation), Genecards (www.genecards.org), Online Mendelian Inheritance in Man (OMIM; http://www.ncbi.nlm.nih.gov/omim), University of California at Santa Cruz genome browser (UCSC; http://genome.ucsc.edu), and UniProt (www.uniprot.org).
Microsatellite Marker Analysis
To investigate the parental origin of the supernumerary ring chromosomes, and to determine whether they originated from a meiosis I or II error, we selected STR markers located inside the chromosomal segments present in the small supernumerary ring chromosomes. For the r(11) these were D11S1395, D11S1920, D11S2005, D11S1313, D11S1357, and D11S4459, for the r(X) these were DXS7132 and DXS6785. Four additional STR markers on chromosome 11, located in the long arm distally from the segment within the r(11), were selected as internal controls (D11S1983, D11S2363, D11S4075, D11S4191). There were no suitable STR markers for the r(12). PCR reactions were performed on 50 ng genomic DNA isolated from peripheral blood. Initial denaturation was at 95°C for 10 min, followed by 33 cycles of denaturation at 95°C for 30 s, annealing at 55°C for 30 s and extension at 72°C for 30 s, and a final extension at 72°C for 30 min. PCR products were analyzed using an ABI 3130 automated sequencer (Applied Biosystems) with LIZ-500 fluorescent size standards and formamide (Applied Biosystems). GeneMapper software (Applied Biosystems) was used for data collection and analysis. The father was unavailable for investigation.
Results
Characterization of sSMCs by Molecular Cytogenetic Methods
Karyotyping of the proband revealed sSMCs (fig. 1) in 98.5% of metaphases from lymphocyte cultures. The number of markers per metaphase varied from 0 to 4 (fig. 1A), with 3 markers being the most frequent number (in 49.5% of metaphases). The markers contained no telomeric DNA as demonstrated by FISH using telomere-specific peptide nucleic acid probes (fig. 1B), and are further referred to as sSMCs. The mother had a normal 46,XX karyotype in all 32 metaphases investigated.

Up to 4 small supernumerary ring chromosomes were found in metaphases of the proband. A Metaphase after Giemsa staining showing 4 small supernumerary marker chromosomes (sSMCs; indicated by arrows). Most metaphases contained 3 or 2 sSMCs. B Three of the sSMCs (indicated by arrowheads) are ring-shaped because they were not labeled by a telomere-specific TTAGGG peptide nucleic acid probe. C Three of the sSMCs originate from chromosomes 11, 12 and X, respectively, as shown by FISH using CEP 11 (locus D11Z1, blue), CEP 12 (locus D12Z3, green) and CEP X (locus DXZ1, red) as probes (all from Abbott-Vysis).
Up to 4 small supernumerary ring chromosomes were found in metaphases of the proband. A Metaphase after Giemsa staining showing 4 small supernumerary marker chromosomes (sSMCs; indicated by arrows). Most metaphases contained 3 or 2 sSMCs. B Three of the sSMCs (indicated by arrowheads) are ring-shaped because they were not labeled by a telomere-specific TTAGGG peptide nucleic acid probe. C Three of the sSMCs originate from chromosomes 11, 12 and X, respectively, as shown by FISH using CEP 11 (locus D11Z1, blue), CEP 12 (locus D12Z3, green) and CEP X (locus DXZ1, red) as probes (all from Abbott-Vysis).
FISH with DNA probes specific for the centromeres of each chromosome demonstrated that 3 small supernumerary ring chromosomes were derived from chromosomes 11, 12, and X (fig. 1C). The origin of the fourth supernumerary chromosome could not be determined because it was present in only 2.5% of metaphases in blood. The analysis of the distribution of the small supernumerary ring chromosomes in cultured and uncultured lymphocytes and uncultured epithelial cells is summarized in table 1. FISH using centromere-specific probes for chromosomes 11, 12 and X showed that 40% of interphase lymphocytes contained these 3 small supernumerary ring chromosomes. In interphase nuclei from urine sediment cells and buccal smears this was 35 and 30%, respectively. The percentages of cells containing different combinations of 2 small supernumerary ring chromosomes was 43–45% in blood and skin, the combination +r(11) and +r(12) being the least frequent in all tissues examined (table 1). In all tissues, the percentage of cells containing no supernumerary ring chromosomes derived from either chromosome 11, 12 or X was low (1.5–5.0%). The small supernumerary r(X) chromosome was present in the highest fraction of cells in all tissues examined (84–89%).

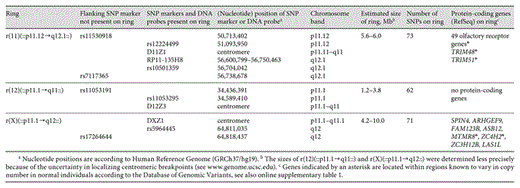
In order to determine the size and the gene content of the ring chromosomes, we initially performed array-CGH using the BAC-array described by Vissers et al. [2003], containing 3,343 BAC clones. This revealed a gain in 8p23.1, identified by a single BAC clone (RP11-80B8) and subsequently confirmed by FISH using RP11-235I5 [Joziasse et al., 2009], as well as a gain in 11q12.1 as indicated by BAC clone RP11-135H8. As a sex-mismatch DNA sample was used as reference for the array-CGH, no copy number gains could be identified for the X chromosome. A higher-resolution investigation was performed using the Illumina Infinium HumanHap317K Genotyping BeadChip. Combining all data from array-CGH, SNP-array and FISH analysis, we conclude that the r(11) contains material from the proximal regions of both the short and the long arm, encompassing 49 olfactory receptor (OR) genes and the genes TRIM48 and TRIM51. The r(12) includes mainly centromeric material, and is devoid of protein-coding genes. The SNP-array demonstrated that the r(X) contains material from the proximal part of the long arm of the X chromosome only, with 8 protein-coding genes. The delineation of the small supernumerary ring chromosomes is summarized in table 2, and the gene content in online supplementary table 1 (www.karger.com?doi=10.1159/000345241). The karyotype of the proband was designated as 46~50,XY,+1~4mar.ish dup(8)(p23.1p23.1)(RP11-235I5++),+r(11)(::p11.12→q12.1::)(D11Z1+),+r(12)(::p11.1→q11::)(D12Z3+),+r(X)(::p11.1→q12::)(DXZ1+).
Parental Origin of the sSMCs
To identify the parental origin of the small supernumerary ring chromosomes and whether they resulted from an error in meiosis I or II, we determined the segregation of SNP and STR markers located on the ring chromosomes of the proband and his mother (summarized in table 3). We tested 6 STR markers located within the segment present in the r(11). For 4 of these the maternal and paternal alleles in the proband could be identified, each showing an increased dosage of the maternal allele in the proband (table 3). We also tested 4 STR markers located more distally on the long arm of chromosome 11 that are not included in the r(11). All of these were informative, showing an equal dosage of the maternally and paternally derived alleles in the proband and his mother (table 3). These observations for the supernumerary r(11) chromosome are consistent with a maternal origin through abnormal segregation during meiosis II. This result is supported by observations on the transmission of STR and SNP markers located on the r(X). The single STR marker tested was heterozygous in the mother of the proband and homozygous in the proband. Five SNP markers located on the r(X) were heterozygous in the mother and homozygous in the proband. This finding is again consistent with a maternal origin of the r(X). Assuming that no recombination occurred between the markers analyzed, the haplotypes can be reconstructed (table 3) to provide the simplest explanation for the segregation of the markers. The SNP-array data also excluded a UPD for either chromosome 11 or 12 in the proband.

Discussion
We report on a 24-year-old man with mild intellectual disability, congenital defects (secundum-type atrial septal defect and a left-sided club foot) as well as obesity. Cytogenetic studies revealed mosaicism for 0–4 sSMCs, 3 of which could be identified as small ring chromosomes derived from chromosomes 11, 12 and X, respectively. Analysis by SNP-array revealed that only the r(11) and r(X) chromosomes contained protein-coding genes.
Since the GATA4 duplication in 8p23.1 does not co-segregate with a cardiac defect in the family of our proband [Joziasse et al., 2009] and because a ∼4-Mb duplication containing GATA4 occurs in about 0.6% of the general population [Zogopoulos et al., 2007], it is unlikely that the additional copy of GATA4 is sufficient to explain the congenital heart defect in our patient. We therefore searched for putative phenotypic effects of the ring chromosomes. To do so, we investigated their gene content and compared our case with published cases with a similar, single sSMC or direct duplication covering the same chromosomal segment (online suppl. table 2). In addition, we determined whether the chromosomal segments within the supernumerary ring chromosomes have been described in the DGV as segmental trisomies occurring in normal, healthy individuals (table 2 and online suppl. table 1). The supernumerary r(11) harbors 49 OR genes as well as the TRIM48 and TRIM51 genes (online suppl. table 1). Based on the presumed gene function and the patterns of transcription of the genes on the r(11) in adult human tissues, there is no apparent relationship to the phenotype of our patient. This is consistent with the existence of several normal individuals carrying an sSMC derived from chromosome 11 of a comparable size (online suppl. table 2, also see the sSMC database compiled by T. Liehr and collaborators (http://www.fish.uniklinikum-jena.de/sSMC.html)). In contrast, individuals with a larger sSMC derived from chromosome 11, containing more material from 11q12.1, show a combination of severe motor delay, hypotonia, seizures and exotropia (sSMC database). In addition, both TRIM48 and TRIM51 are contained entirely within copy number gains detected in normal, healthy subjects (see table 2 and online suppl. table 1). Also CNV of OR genes has been reported, including genes located in the OR cluster in 11q11 [Hasin et al., 2008; Young et al., 2008]. According to the DGV, 28 of the 49 OR genes occur in copy number gains described by several investigators to occur in healthy individuals (online suppl. table 1). The supernumerary r(12) does not harbor protein-coding genes. In addition, the chromosome segment contained in the r(12) has been reported by multiple studies as a copy number gain in healthy individuals (DGV). The sSMC database shows several examples of individuals carrying a single sSMC containing pericentromeric euchromatin from chromosome 12, but who lack an apparent clinical phenotype (see online suppl. table 2). Also, Liehr et al. [2004] concluded from a review of 1,396 published cases that a single sSMC without euchromatic content and without UPD of the structurally normal sister chromosomes does not lead to phenotypic consequences. Based on these observations, it is highly unlikely that the supernumerary r(11) and r(12) chromosomes contribute to the clinical symptoms of our patient. Assuming that the findings in blood are representative for most other tissues and organs, a significant phenotypic effect of the fourth, unidentified sSMC is also unlikely, because in comparison to the other 3 sSMCs, it is present in a much lower percentage of cells (table 1). In addition, it is possible that, similar to the r(12), it does not contain protein-coding genes.
In contrast, the genes on the supernumerary r(X) are more likely to contribute to the phenotype of our patient. Firstly, the r(X) is the most abundant sSMC in both blood (85% of cells) and epithelial cells (84–89% of cells). Therefore, most cells contain 2 copies of the genes on the r(X), in contrast to males with a normal karyotype, who have 1 copy. Secondly, the r(X) does not contain the XIST gene, located in Xq13.2, and therefore it is reasonable to assume that the genes present on the r(X) are transcriptionally active. Thirdly, several patients with an sSMC derived from the X chromosome that contains material from proximal Xq only have been described in the literature (summarized in online suppl. table 2). All of these lack the XIST gene, as shown either by their small size or by lack of hybridization signal with a XIST-specific DNA probe. Although comparison to our patient may be complicated by varying sizes of sSMCs and by different degrees of mosaicism, 9 out of 12 of the previously reported patients have intellectual disability or severe structural brain malformations (online suppl. table 2), similar to our patient. An apparently normal phenotype has been observed in 3 cases. Amiel et al. [1995] described a healthy newborn boy with an X-derived sSMC that was otherwise not characterized in more detail, but that has been detected prenatally in all cultured amniocytes investigated. Starke et al. [2003] described a girl, normal at birth, with a min(X) sSMC that was identified prenatally in 65% of cultured amniocytes. A boy with a small supernumerary r(X) in 25% of blood metaphases had no signs of intellectual disability when observed at 1 year of age [Chen et al., 2006]. In these cases the percentage of cells containing the sSMC may be low, as in the boy described by Chen et al. [2006], or the sSMC may be too small to be of clinical relevance [Amiel et al., 1995; Starke et al., 2003]. We conclude that these observations, together with those on our patient, strongly suggest that supernumerary r(X) chromosomes containing the region between the centromere and the breakpoint in Xq12 are associated with intellectual disability, but only if the degree of mosaicism exceeds a certain, as yet unknown, threshold.
Of the genes on the r(X) that have not been identified in phenotypically neutral copy number gains (see DGV), ARHGEF9 and FAM123B have been associated with hereditary disease (see OMIM). These genes are likely dosage-dependent candidate genes for explaining certain clinical features in our patient. ARHGEF9 (also known as CDC42 guanine nucleotide exchange factor 9) encodes the collybistin protein (CB), a brain-specific GDP/GTP exchange factor, that is essential for clustering of specific GABAA and glycine receptors at inhibitory postsynaptic sites in specific regions of the mammalian central nervous system [reviewed by Papadopoulos and Soykan, 2011]. At inhibitory synapses, CB is required for formation of a hexagonal lattice, built from gephyrin trimers, beneath the postsynaptic plasma membrane [Harvey et al., 2004; Papadopoulos et al., 2007]. In its inactive state CB binds to gephyrin trimers in a 1:1 ratio during microtubule-mediated transport of the trimers to synaptic sites. Observations on CB-deficient mice indicate that CB plays a role in synaptic plasticity and affects the regulation of the excitation/inhibition balance [Papadopoulos et al., 2007, 2008; Jedlicka et al., 2009; Poulopoulos et al., 2009]. In male patients, a missense mutation in ARHGEF9 is associated with hyperekplexia, epilepsy and decline of psychomotor development [Harvey et al., 2004]. A deletion of this gene and a nonsense mutation have been identified in males with intellectual disability and epilepsy [Lesca et al., 2011; Shimojima et al., 2011]. In female patients, balanced translocations that disrupt ARHGEF9 are associated with mild intellectual disability and hyperarousal [Marco et al., 2008], or with severe intellectual disability, epilepsy and anxiety [Kalscheuer et al., 2009]. These findings support the hypothesis that the 2-fold increase in gene dosage of ARHGEF9 in the majority of cells in our patient is causal for the intellectual disability, probably by perturbing the stoichiometry between CB and its interacting proteins [Poot et al., 2011]. We speculate that a 2-fold increase in ARHGEF9 gene dosage leads to excess amounts of gephyrin-free CB molecules that bind to the cytoplasmic NL2 domains, thereby blocking the anchoring points required for gephyrin lattice assembly. However, other possibilities cannot be excluded, such as effects on the relative amounts of CB isoforms that are produced by alternative splicing.
The FAM123B gene has been found to carry somatic loss of function mutations in up to 30% of Wilms’ tumors [Rivera et al., 2007]. Germline mutations cause osteopathia striata with cranial sclerosis [Jenkins et al., 2009]. The gene encodes a component of the β-catenin degradation complex that inhibits WNT signaling and may function in the transcriptional regulation of cellular differentiation [Rivera et al., 2009]. During mouse embryogenesis, the gene is strongly transcribed in the central and peripheral nervous system [Comai et al., 2010]. These observations support a role in intellectual disability as seen in our patient. Because perturbation of WNT signaling has been linked to congenital heart defects in animal models, the additional copy of FAM123B, possibly in conjunction with the GATA4 duplication, may also have contributed to the congenital heart defects seen in our patient [Joziasse et al., 2009]. Contributions from the additional copies of other genes on the r(X) cannot be excluded. For example, the ZC4H2 and ZC3H12B genes both encode zinc-finger proteins, the latter being transcribed in the heart of fetal mice (see the Mouse Genome Informatics database; www.informatics.jax.org).
Both the r(11) and r(X) are present in ∼70 and ∼85% of uncultured peripheral lymphocytes, respectively. This allowed us to determine the segregation of SNP and polymorphic STR markers in order to reconstruct haplotypes for the proband (table 3). Four STR markers on the r(11) were informative. As in each case the maternal allele occurred at a higher dosage than the paternal allele, a maternal meiosis II error is the most likely explanation for the origin of the r(11). Since the father of the proband was not available for investigation, we cannot completely exclude a paternal meiosis I error, a fertilization by 2 sperm cells, which both share the same haplotype for the chromosomal segment within the r(11), or fertilization by a diploid spermatozoon resulting from a paternal meiosis II error. However, these latter scenarios would provide an explanation only if it is further assumed that the father and the mother have an identical haplotype at proximal 11q. Given that there are no indications for consanguinity between the parents, this appears remote. Also, the SNP marker data and 1 STR marker on the r(X) do not support a paternal origin of this sSMC.
In order to explain the occurrence of the sSMCs in our patient, a mechanism must be invoked that causes multiple additional chromosomes to be present in the zygote by misdivision during meiosis II in the fertilized oocyte. As a next step, it must be assumed that the additional chromosomes are incompletely degraded, leading to mitotically stable supernumerary chromosomes that are necessarily small when not interfering with viability. This, then, must happen in such a way that, preferentially, ring chromosomes are formed. First, a model proposing that the multiple sSMCs are the result of fertilization by 2 spermatozoa [Beverstock et al., 2003] is not supported by the DNA marker studies in our patient (although such a scenario may provide an explanation for other patients with a r(Y) as 1 of the multiple sSMCs, see the case described by Weimer et al. [2006]). Another model is based on the incomplete degradation of a non-extruded second polar body during meiosis II [Daniel and Malafiej, 2003]. This mechanism would be compatible with the DNA marker studies in our case. Predivision chromatid nondisjunction can lead to gain of up to 6 chromosomes in the zygote, and may account for the majority of meiosis II errors [Handyside et al., 2012]. We propose that anaphase lagging can explain this. The meiosis II error has to be followed by incomplete degradation of the supernumerary chromosomes, leading to retention of multiple small rings. As demonstrated by array-CGH, about 80% of the non-acrocentric, ring-shaped sSMCs are formed by a break within the centromeric alpha repeat array, as they contain unique-sequence material from 1 chromosome arm only [Baldwin et al., 2008]. This is seen in our patient for the r(12) and the r(X). The other 20% are formed from proximal breaks in both arms [Baldwin et al., 2008], similar to the r(11) in our patient. Array-based investigation of chromosome rearrangements in single cells from cleavage-stage embryos shows that the gain or loss of an entire chromosome arm is the most common type of abnormality during the first mitotic cell divisions [reviewed by Voet et al., 2011], and possibly this is the case also when the multiple sSMCs are generated. These observations are supported by studies of spindle defects and DNA breaks in tumor cells [Martínez-A and van Wely, 2010]. Spindle defects can cause chromosomes to lag behind on the metaphase plate if the kinetochore is attached to both spindle poles. In such lagging chromosomes the opposite tension generated by the microtubules can cause double strand DNA breaks in or adjacent to the centromere, reminiscent of observations of McClintock in maize, leading to a deletion chromosome and a complementary small ring chromosome [Baldwin et al., 2008]. Clearly, these hypotheses need further in-depth studies. In a separate review, we will examine these hypotheses as a putative general model to explain the rare cases of multiple sSMCs, each being derived from a different chromosome.
Acknowledgements
The authors would like to thank the proband and his family members for their participation in this study, and C.G.F. de Kovel for assistance in finding suitable DNA markers. In addition, we are grateful to J.P.M. Geraedts of the Maastricht University Medical Center, Maastricht (The Netherlands), for stimulating discussions and helpful suggestions.
References
R.H. and M.E.v.G. contributed equally to this work.
