

Abstract
The etiology of several autoimmune diseases, including multiple sclerosis (MS), has still not been completely clarified. MS is defined as an autoimmune disease with clinical features of a chronic, inflammatory and demyelinating autoimmune disorder which affects the central nervous system. The course of the disease includes phases of remission and relapses which can be exacerbated in both severity and duration. Chemokines, which are a subfamily of the cytokines, act as chemoattractants for a wide variety of cells, including immune cells. CXCL10 is a small protein that is defined as an ‘inflammatory' chemokine and binds to CXCR3 to mediate immune responses through the activation and recruitment of leukocytes such as T cells, eosinophils, monocytes and NK cells. The aim of this review is to address recent findings regarding the relationship between CXCL10 and MS.
Introduction
To date, the etiological aspects of many autoimmune diseases, including multiple sclerosis (MS), has yet to be characterized completely [1]. MS is an inflammatory disorder of the central nervous system (CNS) which is defined by loss of myelin, gliosis and different degrees of axonal and oligodendrocyte pathology [2]. MS usually commences in early adulthood, with paralysis, sensory disturbances, lack of coordination and visual impairment among its most frequent features [3]. The disease often begins with an ‘attack' or ‘flare' that lasts from a few days to weeks and is followed by remission that most often lasts from a few months to years [4,5]. The variable course of the disease may be reflected in the differences seen at the morphological level by magnetic resonance imaging and histopathologic evaluations of the CNS, which also vary significantly [6]. Although the main causes of MS are yet to be fully understood, the disease mechanisms are often inferred from the effector phase of an animal model of MS: experimental autoimmune encephalomyelitis (EAE) [7]. The disease course of relapsing-remitting MS (RRMS) involves periods of clinical remission and unpredictable relapses or exacerbations, differing in both severity and duration. Following several years of an RR phase, MS patients may enter a secondary progressive phase (SPMS) where the onset of symptoms and nerve functions gradually worsen either with or without relapses [8].
Although the underlying etiological cause and pathogenesis of MS are unknown, the current suggestions favor MS as an autoimmune inflammatory disorder of the CNS in which autoreactive T lymphocytes recognize CNS-specific proteins, leading to inflammation and demyelination. According to the cellular infiltrates detected in the brain and cerebrospinal fluid (CSF) from patients alongside data from rodent models, including EAE, MS is largely considered to be a CD4+ T helper 1 (Th1)-mediated inflammatory disease [9]; however, CD4+ Th17 effector T cells are postulated to have more important roles [10,11]. In the EAE model, it has been demonstrated that the injection of myelin components into susceptible animals resulted in a CD4+-mediated autoimmune disease that shares similarities with MS [12]. EAE can also be induced by the adoptive transfer of encephalitogenic CD4+ T cells into a naive animal [12,13]. It appears that white blood cells, including lymphocytes, are chemoattracted to the MS plaques in response to chemokines. Immunohistochemical studies demonstrated that several chemokine ligands, in parallel with their receptors, are present in MS lesions. Therefore, it seems that chemokines play crucial roles in the pathogenesis of MS. Chemokines are a subfamily of the larger family of cytokines that serve as recruiter/migratory factors for a wide spectrum of cells, and their target cells express appropriate transmembrane G protein chemokine receptors. Chemokines are further subdivided into C, CC, CX3C and CXC subgroups according to the position of conserved cysteine motifs in their structure [14]. CXCL10 (interferon-γ-inducible protein 10, previously called IP-10) was initially discovered as a chemokine which is induced by interferon (IFN)-γ and is produced by a wide range of cell types including monocytes [15], neutrophils [16], endothelial cells [17], keratinocytes [18], fibroblasts [19], mesenchymal cells [20], dendritic cells [21], hepatocytes [22] and astrocytes [23]. CXCL10 binds to CXCR3, which is discussed in the next section. Recent evidence has indicated that serum/tissue expression of CXCL10 is increased in MS [24]. Both CXCL10 and CXCR3 are crucial for leukocyte trafficking and homing to inflamed tissues as well as the perpetuation of inflammation which leads to tissue damage [25].
Th1 and Th2 subsets of lymphocytes can be defined by their expression of chemokine receptors. CXCR3 is associated with the Th1 phenotype and is preferentially expressed on activated Th1 cells [26]. MS is considered a Th1-dependent disease in which Th1-related cytokines and chemokines are increased during the course of the disorder [27]. Interestingly, elevated percentages of T cells expressing the CXCR3 chemokine receptor in peripheral blood and CSF during the active phase of MS suggest that CXCR3 plays important roles in the induction of active demyelinating in MS brain lesions [28,29]. Based on the clinical and experimental observations discussed above, this review explores the relationship between CXCL10 and MS as an autoimmune disorder.
CXCL10 Biostructure and Functions
It is well documented that chemokines - and more specifically CXCL10 - are produced by several cell and tissue types and exhibit pleiotropic effects on a wide range of biological processes including immunity, angiogenesis and organ-specific metastasis of cancers. The role of CXCL10 in these processes makes it a promising therapeutic target for various diseases. However, an identification of the structural properties and mechanism of action of CXCL10 needs to be made if CXCL10 is to be used as a worthwhile therapeutic target for treatment of the aforementioned pathological conditions. CXCL10 was initially identified in human U937 cells (a histiocytic lymphoma cell line with monocytic characterization and origin) and from human placenta and spleen as an IFN-γ-inducible product [30]; mob-1 and crg-2 are the rat and mouse homologs of human chemokine CXCL10, and they share 70 and 78% amino acid homology with these proteins, respectively [31]. Like other members of the chemokine subfamily, CXCL10 is a small-molecular-weight protein (10 kDa) which was functionally described as an ‘inflammatory' chemokine. Furthermore, the lack the ELR tripeptide (Arg-Leu-Glu) motif in the vicinity of CXC residues characterizes CXCL10 as an inhibitor of neovascularization; hence, it acts as an ‘angiostatic' (antiangiogenic) chemokine [32]. The CXCL10 gene is located on chromosome 4, contains 4 exons and 3 introns and encodes a protein of 98 amino acids [19,33]. CXCL10 is transcriptionally regulated in response to external stimuli such as IFN-γ and lipopolysaccharides (LPS) by a region of 230 nucleotides upstream from the transcriptional start site. This area contains several important regulatory elements, as follows: (a) 2 sites for nuclear factor-κB (NF-κB); (b) 1 site for activator protein 1; (c) 1 site for interferon-stimulated response element (ISRE); and (d) 1 site for binding of heat shock (HS) factors (fig. 1) [34].
![Fig. 1. External stimuli which affect CXCL10 and the intracellular events leading to its expression (adopted from Ahmadi et al. [30]). Signals are initiated by receptor-ligand interaction, either involving growth factors or cytokines, and stress stimuli are illustrated. Process of receptor binding. Protein complexes are recruited to the receptor. Resulting activation of corresponding signaling events and initiation of activation of their downstream targets. CS = Cytokine stimulation; CRE = cytokine response element; HSRE = HS response element; US = unknown stimulation; ERK = extracellular signal-regulated kinase; IκB = inhibitor of NF-κB; JNK = c-Jun N-terminal kinase; MEK = mitogen-activated protein kinase kinase; NIK = NF-κB-inducing kinase; PI3K = phosphoinositide 3-kinase; UV = ultra violet light.](https://karger.silverchair-cdn.com/karger/content_public/journal/nim/21/6/10.1159_000357780/2/m_000357780_f01.jpeg?Expires=1716292387&Signature=0Bn3ZCURBdmwxdqAfwfTjDEkeaXe~7wJPB-39XF8z7Uza7HIo7KlALWa63-S9ceN5kmIerEIrZr-~Sd4xFFNqoR8ghZMvzUwkmKyUvu0W81EQEYVj8GLcwEVPa5xOg7y1xmK17p4no5Ql5Q9BCWwvsJ~vd06-Trr3bZlzMUcTt6occegaVKe~cqST4hvZypqhbyyrJENLfTrgcsWLOAHHIgSR~FfvSQTzU3QltQxUR9Muz8al5~U6zQ53eYQga1GV5yQIp3sifH13riK8ICLtcqIrGoB83QlsuJOhn4uDuoUka2yNe672sNd4dvs1zk6Y4Hkhdv1KGOjmeR-a5FiXQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
External stimuli which affect CXCL10 and the intracellular events leading to its expression (adopted from Ahmadi et al. [30]). Signals are initiated by receptor-ligand interaction, either involving growth factors or cytokines, and stress stimuli are illustrated. Process of receptor binding. Protein complexes are recruited to the receptor. Resulting activation of corresponding signaling events and initiation of activation of their downstream targets. CS = Cytokine stimulation; CRE = cytokine response element; HSRE = HS response element; US = unknown stimulation; ERK = extracellular signal-regulated kinase; IκB = inhibitor of NF-κB; JNK = c-Jun N-terminal kinase; MEK = mitogen-activated protein kinase kinase; NIK = NF-κB-inducing kinase; PI3K = phosphoinositide 3-kinase; UV = ultra violet light.
External stimuli which affect CXCL10 and the intracellular events leading to its expression (adopted from Ahmadi et al. [30]). Signals are initiated by receptor-ligand interaction, either involving growth factors or cytokines, and stress stimuli are illustrated. Process of receptor binding. Protein complexes are recruited to the receptor. Resulting activation of corresponding signaling events and initiation of activation of their downstream targets. CS = Cytokine stimulation; CRE = cytokine response element; HSRE = HS response element; US = unknown stimulation; ERK = extracellular signal-regulated kinase; IκB = inhibitor of NF-κB; JNK = c-Jun N-terminal kinase; MEK = mitogen-activated protein kinase kinase; NIK = NF-κB-inducing kinase; PI3K = phosphoinositide 3-kinase; UV = ultra violet light.
CXCL9, CXCL10 and CXCL11 all share CXCR3 as a common receptor for their activities. CXCR3 has two different isoforms: CXCR3-A and CXCR3-B. The most recent studies revealed that the CXCR3 isoforms differentially regulate cell function. The CXCL10/CXCR3-A axis contributes to the induction of chemotaxis and proliferation in various cell types [35,36], whereas the CXCL10/CXCR3-B axis inhibits migration and proliferation but induces apoptosis [36,37]. Additionally, CXCL10 is reported to act as an antiangiogenic/antitumor protein [37,38]. Although the precise biological functions of CXCL10 are yet to be completely identified, several lines of evidence have demonstrated that this chemokine is involved in the following physiological and pathological situations: (a) chemoattraction of macrophages, monocytes and activated T and NK cells [39,40,41]; (b) modulation of T cell development and function [42,43]; (c) inhibition of in vitro colony formation by early human bone marrow progenitor cells [44]; (d) stimulation of T cell adhesion to endothelial cells [45]; (e) induction of NK cell migration along with NK cell-mediated cytolysis; (f) in antiangiogenesis (including antitumor angiogenesis) [24,30,46], and (g) by mitogenic and chemotactic effects on vascular smooth muscle cells [43,44].
CXCL10 Signal Transduction
Synergy was observed between tumor necrosis factor (TNF)-α and IFN-γ regarding the induction of CXCL10 expression by different cell systems such as keratinocytes and hepatocytes. This synergistic effect is facilitated by NF-κB and ISRE in the promoter of CXCL10 [47,48]. In isolated and cultured pancreatic acinar cells, cholecystokinin (CCK8) was shown to increase CXCL10 via a pathway regulated by NF-κB activation which was blocked by PDTC (pyrrolidine dithiocarbamate, an NF-κB inhibitor) through a mechanism which repressed inhibitor of NF-κB-α degradation and in turn caused induced CXCL10 expression [49]. NF-κB activation and subsequent CXCL10 expression, in response to CCK8, was reported to be mediated by protein kinase C in parallel with increased intracellular Ca2+ [49,50]. Evidence also indicates that stimulation of keratinocytes with TNF-α and IFN-γ induced CXCL10 in a concentration- and time-dependent manner, mediated by activation of the protein kinase C pathway [47,51]. In a similar fashion, primary human kidney mesangial cells, stimulated by TNF-α and/or IFN-γ, also produced CXCL10 in an NF-κB-dependent pathway, and co-operation between the ISRE and NF-κB sites on the CXCL10 promoter were responsible for this action [52]. TNF-α and IFN-γ employ STAT1α (signal transducer and activator of transcription 1α) and NF-κB, respectively, for CXCL10 expression in human fibroblast cell lines [47,52,53] as well as LPS-stimulated Kupffer cells [54].
Role of CXCL10 in MS
Currently there is limited information available concerning the role of chemokines in MS, especially at the serum level. In MS, chemokines act both as leukocyte-migratory factors and mediators of proinflammatory reactions (both of which promote the process of demyelination) [55]. Chemokines regulate the expression of adhesion molecules and therefore aid transendothelial migration of autoreactive immune-compatible cells through the blood-brain barrier [56].
Serum and CSF Levels of CXCL10 in MS Patients
The levels of CXCL10 in circulating blood were previously found to be elevated in IFN-β-treated MS patients when compared with those left untreated [57]. Altered serum levels of other members of the chemokine family such as CCL11 indicated that they are not directly associated with IFN-β treatment and are regulated by Th2 cell-mediated cytokines [58].
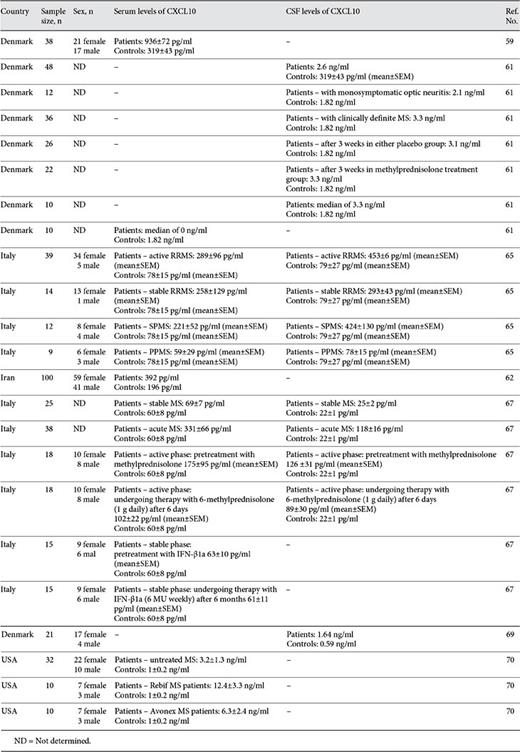
Sørensen et al. [59] reported that CXCL10 levels were higher in CSF specimens collected from patients suffering from active MS as opposed to CSF measurements made in neurological subjects with noninflammatory conditions. More than 90% of the T cells present in the CSF of MS patients expressed CXCR3, which is significantly more than those found in peripheral blood. Present observations are in accordance with the fundamental role of the CXCL10/CXCR3 axis in the pathogenesis of MS [29,59,60].
Sørensen et al. [61] in another study also revealed that CXCL10 levels were elevated in the CSF of MS patients and that this was associated with increased leukocyte numbers in CSF [61,62]. It could be reasonable to assume that the induction of CXCL10 (especially in RRMS patients) is probably related to stimulation of regulatory motifs present at the upstream transcriptional start site of the CXCL10 promoter. This regulatory region of the CXCL10 gene contains motifs for a variety of signal transduction pathways including HS IFN-γ and NF-κB response elements [63,64 ](fig. 1) [30].
Scarpini et al. [65] assessed CXCL10 in both CSF and serum of RRMS and SPMS patients, but not in primary progressive (PP) MS patients, and observed its elevation. Similarly, Sprenger et al. [66] indicated that CXCL10 was significantly enhanced in the CSF and serum of patients affected by other inflammatory neurological diseases. The results from all these studies are in line with data collected by Franciotta et al. [67] as well as our own data [62], which showed elevated serum levels of CXCL10 in RRMS patients. The increased CXCL10 levels and the increase in cells expressing its receptor, CXCR3, in the CSF of MS patients during the active phase of the disease in the intrathecal compartment have also been well documented [59]. Additionally, Comini-Frota et al. [68] reported that serum CXCL10 levels were higher in MS patients than in controls, and this is interesting because they also recorded considerable increases in expression following 36 h of IFN-β1a or IFN-β1b therapy.
Again, studies showed that CXCL10 expression was elevated in the CSF of MS patients and this was associated with demyelination in CNS tissue sections. Furthermore, this was tightly correlated with CXCR3 expression [69]. Christophi et al. [70] reported that quantities of CXCL10 and caspase 1 were both elevated in peripheral blood mononuclear cells of MS patients. However, treatment with either IFN-β1a formulations (Rebif or Avonex) significantly upregulated the expression of both CXCL10 and caspase 1, which was mediated by the activation of the STAT1 pathway. Moreover, they observed that CXCL10 expression (at the mRNA level) was induced more by the use of Rebif than Avonex. Therefore it appears that in addition to IFN-γ, TNF-α and proinflammatory cytokines CXCL10 is also an IFN-β-inducible gene product involved in MS pathogenesis [61,71,72]; however, conflicting reports claimed that chemokine expression was also suppressed by IFN-β [73].
In contrast to the aforementioned studies which documented the elevation of CXCL10 levels in MS patients, Sørensen et al. [61] reported that CXCL10 remained significantly unchanged within the CSF 1 week following fulfillment of a 15-day course of oral high-dose methylprednisolone in MS patients. This does not exclude an effect during or immediately after treatment. However, other effects of methylprednisolone appear to be related to the properties of this compound during the processes of disease remission observed after treatment [74,75]. However, the current observations are in agreement with the prominent role of CXCL10 in the maintenance of intrathecal inflammation and may partly explain the mechanism(s) by which high doses of oral methylprednisolone do not influence the recurrence of disease activity [74]. Thus, treatment with IFN-β appears to be the preferred regime leading to decreased intrathecal inflammation [76].
More recent reports by Mellergård et al. [77] verified that following a period of 1 year of treatment with the newly introduced anti-MS reagent natalizumab, a panel of cytokines and chemokines were decreased. They demonstrated that, in addition to declined levels of proinflammatory cytokines such as interleukin (IL)-1β, IL-2, IL-4, IL-5, IL-6, IL-8, IL-10, TNF-α, IFN-δ and granulocyte/macrophage colony-stimulating factor, several chemokines including CXCL9, CXCL10, CXCL11, CCL17 and CCL22 were also decreased in the blood and CSF of RRMS patients following 1 year of treatment with natalizumab.
Expression of CXCL10 in EAE Animal Models and MS Lesions
Astrocytes are also the source of CXCL10 in both human and mouse EAE models [29,59,78], and evidence from these models demonstrates that both TNF-α and IFN-γ (as members of the proinflammatory cytokines) regulate CXCL10 expression in cell lines [79] as well as a rat model [48].
Immunocytochemical studies confirmed the presence of CXCL10 in postmortem CNS tissues from MS patients. In lesions where demyelination was active and ongoing, CXCL10 was predominantly expressed by both macrophages (present inside the plaque) and reactive astrocytes in the surrounding parenchyma. Correspondingly, CXCR3 was also expressed by T cells and astrocytes within the plaques, which may strongly suggest an autocrine fashion of CXCL10 expression by these cells [29,60].
Salmaggi et al. [80] demonstrated that human brain microvascular endothelial cells and astrocytes express IFN-γ-inducible chemokines CXCL10 and CXCL11 in response to inflammatory stimuli. Moreover, in autoimmune pathologies including MS, both δ T cells (Vδ1 and Vδ2 cells) exhibit differential expression of cell adhesion molecules as well as chemokine receptors. Vδ1 cells express CXCR4, while Vδ2-type cells express CXCR3, and these cell populations transmigrate across endothelial cells in response to CXCL12 and CXCL10 [81]. Buttmann et al. [82] investigated a panel of IFN-inducible chemokines including CXCL10 and, using in situ hybridization analysis, corroborated a strong expression of CXCL10 mRNA by infiltrating immune cells and basal keratinocytes into patients in the IFN-β-injected areas. Table 1 shows a summary of the literature reviewed concerning the immune-related diseases reported.
Conclusions
Overall, based on the latest information available in the literature regarding the role of CXCL10 in the pathogenesis of MS, the authors of the present article propose that CXCL10 plays a critical role during symptomatic inflammatory demyelination events that occur in the pathogenesis of MS. However, this is inconsistent with the evidence that exists regarding the CSF and serum levels of CXCL10 in the PP form of MS, in which less inflammation is observed [83]. Indeed this is more similar to what is observed in noninflammatory neurological diseases.
Finally, due to the complex manifestations of MS, more characterization - with particular attention to accuracy - is required with regard to surveying several other aspects of the disease. To achieve this, the authors of the present article believe that it is worthwhile to more accurately examine the role of CXCL10 and its corresponding receptor (e.g. CXCR3) during the pathogenesis of MS using animal-based EAE models.
Acknowledgement
This project was supported by a grant from the Rafsanjan University of Medical Sciences.
Disclosure Statement
None of the authors of this study declare any conflict of interest.
