

Abstract
Background: Chronic administration of <smlcap>D</smlcap>-galactose (<smlcap>D</smlcap>-gal) results in oxidative stress and chronic inflammatory aging. Age-related changes in the brain result in neurovascular damage and blood-brain barrier (BBB) dysfunction. However, little is known regarding <smlcap>D</smlcap>-gal-induced neurovascular damage, as well as the protective effects of huperzine A. Objective: The purpose of this study was to utilize a<smlcap> D</smlcap>-gal-induced rat model to investigate the activation of neurovascular inflammatory damage and apoptosis in the rat hippocampus and to understand whether huperzine A alleviates <smlcap>D</smlcap>-gal-induced neuronal and vascular inflammatory injury. Methods: Aging rats were treated with <smlcap>D</smlcap>-gal (300 mg/kg s.c. for 8 weeks), were coadministered <smlcap>D</smlcap>-gal and huperzine A (<smlcap>D</smlcap>-gal 300 mg/kg and huperzine A 0.1 mg/kg s.c. for 8 weeks) or served as the saline-treated control group rats (same volume of saline given subcutaneously for 8 weeks). Changes in hippocampal morphology and biomarkers of inflammatory damage were analyzed. Results: Our study revealed that chronic administration of <smlcap>D</smlcap>-gal resulted in the activation of glia and vascular endothelial cells and upregulation of mRNA and protein levels of cell-associated adhesion molecules and inflammatory cytokines via nuclear factor (NF)-κB inhibitor degradation and NF-κB nuclear translocation. The inflammatory injury caused significant BBB dysfunction, decreased density of tight junctions (TJs) and apoptosis in the rat hippocampus. Coadministration of huperzine A not only markedly inhibited the <smlcap>D</smlcap>-gal-induced increase in acetylcholinesterase (AChE) activity, but also alleviated <smlcap>D</smlcap>-gal-induced neurovascular damage by inhibiting <smlcap>D</smlcap>-gal-induced NF-κB activation, improving cerebrovascular function and suppressing the <smlcap>D</smlcap>-gal-induced decrease in the density and protein levels of TJs and cell apoptosis. Conclusions: Our findings provided evidence that <smlcap>D</smlcap>-gal induced a proinflammatory phenotype mediated by NF-κB in the rat hippocampus. Moreover, huperzine A suppressed <smlcap>D</smlcap>-gal-induced neurovascular damage and BBB dysfunction, partly by preventing NF-κB nuclear translocation. The inhibiting effect of huperzine A on AChE activity might play an important role in attenuating <smlcap>D</smlcap>-gal-induced inflammatory damage.
Introduction
Chronic administration of D-galactose (D-gal) which induces neurotoxicity is a common model used to study aging and memory impairment [1, 2, 3, 4, 5]. There are 3 main aspects of the mechanism of D-gal-induced aging. First, the abnormal product of D-gal metabolism, galactitol, accumulates in the cell and causes the production of reactive oxygen species (ROS) [6]. Excessive ROS exposure results in the accumulation of somatic mutations of mitochondrial DNA and the production of damage-associated molecular patterns (DAMPs). The mitochondrial DNA mutations are transmitted during mitochondrial and cell division. The impairment of mitochondrial DNA function causes further ROS production [6, 7]. Consequently, D-gal-induced DAMPs lead to inflammatory aging and immunosenescence via activation of inflammatory pathways [8, 9, 10]. Second, D-gal reacts with free amine groups of amino acids within proteins and peptides to form advanced glycation end products (AGEs) [11]. These products in turn activate the receptor for AGEs to mediate sterile inflammation [12]. Third, the aged and demented nervous system exhibits significant cholinergic dysfunction [13]. Likewise, chronic administration of D-gal in mice results in remarkable increases in acetylcholinesterase (AChE) [14]. Thus, the DAMPs released during conditions of oxidative stress might mediate D-gal-induced sterile inflammation via pattern recognition receptors (PRRs) and non-PRRs. Inhibition of the cholinergic anti-inflammation pathway exaggerates D-gal-induced inflammatory damage. Chronic and systemic sterile inflammation also leads to tissue destruction and disease [12].
Neurons, glia, pericytes and endothelial cells of the cerebral microvasculature coexist in an intimate proximity in nervous tissues. These cells become activated and can be coordinated in the defense against inflammatory stimuli, such as chronic systemic D-gal exposure, traumatic brain injury and focal cerebral ischemia [15, 16, 17]. Inflammation plays a critical role in the host defense against invasive pathogens and DAMPs. Glia and endothelial cells are activated in the neurovascular unit during inflammation and express cell-associated adhesion molecules, such as E-selectin, intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1) and chemokines, which facilitate the rolling, adhesion, activation and migration of circulating leukocytes across the endothelial cell barrier to the site of inflammation [18, 19, 20]. Excessive leukocyte accumulation during inflammation mediated by the overexpression (or sustained expression) of adhesion molecules can damage neurovascular tissues, including cell death and deterioration of the blood-brain barrier (BBB). For example, postischemic microglial activation exaggerates the injury to the BBB. The BBB consists of highly specialized endothelial cells, which separate the brain from the peripheral blood with very limited permeability. To maintain homeostasis, various materials selectively move across the plasma membrane and tight junctions (TJs) via transcellular and paracellular pathways, respectively [21]. Compared to the endothelial cells of nonneural tissues, one component of the difference in the vascular permeability may be related to the expression levels of the major constituents of TJ strands (e.g. claudin-5), plasma membrane spanning proteins (e.g. occludin) and scaffold cytoplasmic proteins (e.g. zonula occludens-1, Zo-1) [22, 23, 24, 25]. The permeability and TJ integrity of the BBB are significantly altered in animal models and patients with Alzheimer's disease [26, 27, 28, 29]. Deficiency of claudin-5, a major constituent of TJ strands in brain endothelial cells, results in size-selective relaxation of the BBB [23]. During the aging process, oxidative stress leads to inflammatory damage and dysfunction of the BBB [30, 31]. To the best of our knowledge, little is known regarding D-gal-induced neurovascular inflammation and alteration of TJs and BBB function.
Huperzine A is a reversible and selective AChE inhibitor (AChEI) that exhibits neuroprotective effects by suppressing the activation of glial cells and overexpression of proinflammatory factors, as demonstrated in a rat model of transient focal cerebral ischemia [19]. AChEIs can induce cholinergic upregulation with subsequent effects on neuroinflammation and hepatic inflammation, such as hepatic and T-cell proliferation, glial activation and cytokine production [10, 20, 32]. Huperzine A has been widely used to improve cognitive and memory deficits in patients with benign senescent forgetfulness, Alzheimer's disease and vascular dementia via noncholinergic mechanisms, including attenuating oxidative stress, inhibiting apoptosis and interfering with amyloid precursor protein metabolism [33, 34, 35, 36]. In addition, huperzine A may protect against diverse neurodegenerative diseases by blocking N-methyl-D-aspartate receptors [37]. Hippocampal dysfunction, such as cognitive and memory deficits, is closely associated with neurodegenerative disease [15, 20]. In this study, we chose the hippocampus to investigate whether D-gal-induced inflammatory injury to the BBB and the neurovascular unit via the nuclear factor κB (NF-κB) inflammatory pathway might contribute to central nervous system aging. We report that huperzine A-mediated attenuation of D-gal-induced aging occurs, in part, due to inhibition of D-gal-induced neuroinflammation, as well as decreased deterioration and dysfunction of the BBB, via the NF-κB inflammatory pathway.
Materials and Methods
Animals and Drug Administration
Male Sprague-Dawley rats (200-220 g, Laboratory Animal Center, Shanghai Medical College, Fudan University) were randomly separated into 3 groups: (1) D-gal-treated group (n = 15), (2) D-gal- and huperzine A-treated group (n = 15), and (3) the saline-treated control group (n = 15). The D-gal-treated group was subcutaneously injected with 300 mg/kg D-gal daily for 8 weeks; the D-gal- and huperzine A-treated group was subcutaneously coinjected with 300 mg/kg D-gal and 0.1 mg/kg huperzine A daily for 8 weeks, and the saline-treated control group was injected with saline. Experimental animals were recruited into 2 independent trials. In the second trial, 3 rats in each group were used for the AChE activity assay and claudin-5 semiquantitative Western blot analysis. The Animal Care and Use Committee of the Shanghai Medical College of Fudan University approved all of the animal procedures.
Tissue Preparation
The animals (n = 6 in each group) were sacrificed with an overdose of sodium pentobarbital and transcardially perfused with 4% paraformaldehyde solution 24 h after the last drug administration. The brains (n = 3) were postfixed overnight in 4% paraformaldehyde and were cryoprotected using a 30% sucrose solution. Coronal sections of 50 µm thickness were cut through the hippocampus using a cryostat. The hippocampus (n = 3) was then postfixed with 1% osmium tetroxide, 1.5% ferrocyanide for 2 h in the dark, dehydrated and embedded in Epon LX 112 resin. Ultrathin sections were stained with uranyl acetate and lead citrate, and the sections were then examined using electron microscopy.
Immunohistochemistry
For glial fibrillary acidic protein (GFAP), E-selectin and CD11b immunohistochemistry, the sections were first treated with 3% hydrogen peroxide for 10 min at room temperature (RT), followed by rinsing thoroughly in phosphate-buffered saline (PBS). Next, the sections were washed in PBS, blocked and then incubated in mouse monoclonal GFAP, rabbit polyclonal E-selectin antibodies (Santa Cruz Biotechnology Inc., Dallas, Tex., USA) and rabbit polyclonal CD11b antibody (Abcam, Cambridge, UK) at RT. The sections were washed 3 times in PBS, incubated for 1 h in a secondary antibody solution of biotinylated goat anti-mouse IgG (Beyotime Institute of Biotechnology, Suzhou, Jiangsu, PR China), washed 3 times and treated with horseradish peroxidase-labeled streptavidin for 1 h. The immunohistochemical reaction products were visualized using a Diaminobenzidine Horseradish Peroxidase Color Development Kit (Beyotime Institute of Biotechnology). Hematoxylin was used to stain cellular nuclei. The negative saline-treated control sections for GFAP, E-selectin and CD11b antigen were processed in the same manner except that the primary antibody incubation step was replaced by continued incubation in normal serum. Neither any positive immunostaining nor any recognizable background staining was observed in the negative saline-treated control sections.
Transferase-Mediated Uridine Nick End Labeling Assay and Immunofluorescence
DNA strand breaks in the hippocampal sections were labeled by terminal deoxynucleotidyl transferase with the transferase-mediated uridine nick end labeling (TUNEL) labeling mixture (Roche Applied Science, Mannheim, Germany). After 2 washes with PBS, 50 μl of the TUNEL reaction mixture was added to the sample. The negative saline-treated controls were made by adding 50 μl of the labeling solution instead of the TUNEL mixture. The samples were incubated in a 37°C incubator for 60 min, and the reaction was terminated with 3 PBS washes. The sections were further incubated for 1 h at RT with PBS containing 1% (w/v) bovine serum albumin, 5% heat-inactivated goat serum and 1% Triton X-100 in 0.1 M PBS and then incubated at 4°C for 48 h in rat endothelial cell antigen-1 (RECA-1) antibody (Santa Cruz Biotechnology) solution [in PBS-T1 containing 1% (w/v) bovine serum albumin, 5% heat-inactivated goat serum and 0.1% Triton X-100 in 0.1 M PBS]. The slides were washed in PBS 3 times and then exposed to the secondary antibody, goat anti-mouse IgG1-TITC (1:100), for 4 h at RT. After several washes, the tissues were mounted onto glass slides, coated with glycerin, coverslipped and then subsequently stored at 4°C. To the saline-treated control group for secondary antibody nonspecificity, the sections were processed with buffer in which the primary antibody was excluded. In this case, only dim autofluorescence was observed, with no signal similar to that observed with the primary antibody when viewed under a fluorescence microscope (Leica, Solms, Germany).
Immunoglobulin G Staining for BBB Evaluation
BBB integrity was assessed by measuring the level of immunoglobulin G (IgG) extravasation into the brain using previously described methods [38, 39]. Briefly, coronal brain sections with the CA3 region of rat hippocampus were incubated for 4 h at RT using a biotin-conjugated affinity-purified rabbit anti-rat IgG antibody (1:1,000, Santa Cruz, Calif., USA) diluted in PBS containing 0.1% Triton X-100 and 1% bovine serum albumin. After several washes, the sections were incubated for 1 h at RT with 4′,6-diamidino-2-phenylindole, and the fluorescence intensity was quantified using a fluorescence microscope (Leica, Germany) with an excitation wavelength of 494 nm and an emission wavelength of 525 nm. The image was then converted into the average integrated intensities using the Leica DFC320CCD image analysis software.
Quantitative Reverse Transcription Polymerase Chain Reaction Analysis of mRNA Expression
RNA was isolated from the hippocampal tissues of 3 rats using Trizol® reagent (Invitrogen, Carlsbad, Calif., USA). Isolated total RNA (5 µg) with an absorbance ratio of 1.8-2.0 was reverse-transcribed into cDNA using SuperScript® III reverse transcriptase (Invitrogen). The cDNA template (1 µl) was amplified using SYBR® Green (Invitrogen) DNA-binding dye for quantitative polymerase chain reaction. The thermal cycling conditions were as follows: 2 min initiation at 95°C, followed by 40 amplification cycles (10 s at 95°C, 30 s at 60°C and 45 s at 70°C). The melting temperature was 70-95°C. The mRNA levels were normalized to the reference gene β-actin and calculated as 2-ΔCT[40]. We performed 3 trials, each with 3 repetitions. The 3 CT values and the 2-ΔCT value were obtained for each trial. The primer sequences are shown in table 1.
Primers for quantitative real-time polymerase chain reaction

Semiquantitative Western Blot Analysis
The rat hippocampal tissues were homogenized using an ultrasonic cell disruptor in ice-cold lysis buffer with Halt™ protease and phosphatase inhibitor single-use cocktail (Pierce Thermo Scientific, Rockford, Ill., USA). The lysates were centrifuged at 10,000 g for 10 min at 4°C, and the supernatant solutions were collected. The total protein concentration of each sample was determined using a protein assay kit (BCA kit, Pierce). For p65 detection, cytoplasmic and nuclear extracts were prepared using a nuclear and cytoplasmic protein extraction kit (Beyotime Institute of Biotechnology) according to the manufacturer's instructions. The lysates were separated by electrophoresis and transferred onto polyvinylidene difluoride membranes using a wet transfer system (Bio-Rad Laboratories Inc., Hercules, Calif., USA). The membranes were incubated for 1 h at RT in 5% (w/v) milk in PBS with 0.1% Tween-20 and probed with primary antibodies to claudin-5 (Bioss, Freiburg, Germany), NF-κB-p65 (p65), p-NF-κB-p65 (phospho-p65), interleukin (IL)-6, NF-κB inhibitor (IκBα; Santa Cruz Biotechnology, Santa Cruz, Calif., USA), tumor necrosis factor (TNF)-α, IL-1β (Abcam, UK), E-selectin, ICAM-1, VCAM-1, CD11b+, active caspase-3, occludin, Zo-1 (Boster, PR China), glyceraldehyde 3-phosphate dehydrogenase (GAPDH), histone H3 (His) and β-actin (Beyotime), followed by incubation with horseradish peroxidase-conjugated goat anti-rabbit or anti-mouse IgG and detection using a chemiluminescence substrate (ECL plus, Amersham Pharmacia Biotech, UK), viewed in a Molecular Imager® (Bio-Rad). The optical density values of the target proteins detected in each experimental group were normalized to the optical density values of the corresponding saline-treated control groups, such as GAPDH, His and β-actin.
AChE Activity Assay
The spectrophotometric assays were performed with a Shimadzu UV-2101PC scanning spectrophotometer equipped with a thermostable cell compartment and cell positioner. All of the fluorometric assays were performed on a Hitachi F-2000 fluorescence spectrophotometer.
After weighing, each cerebral cortex or hippocampus was homogenized in cold 0.1 M PBS (pH = 7.4) in a Tissuelyser-24 tissue grinder on ice. The ratio (w/v) of each sample to PBS was 100 mg sample per 0.9 ml PBS. Then, the samples were centrifuged at 10,000 g for 10 min at 4°C. The protein concentrations of the supernatant solutions were determined using a BCA Assay Kit (Beyotime). For the AChE activity assay, hydrolysis of 1 µmol of substrate in the response system was defined as 1 activity unit. Thirty microliters of the supernatant solution were used in the AChE Assay Kit (Nanjing Jianchen Bio, Jiansu, PR China), and acetylthiocholine chloride was used as the substrate. The AChE activity of the supernatant solutions was determined by spectrophotometry. The experiment was performed according to the protocol in the assay kit.
Data Analysis
All of the data are presented as the mean ± SEM. The data from the 3 experimental groups (D-gal-treated group, D-gal and huperzine A-treated group, and the saline-treated control group) were analyzed together using 1-way ANOVA followed by the Holm-Sidak method for all pairwise multiple comparison procedures. Differences with a p value <0.05 were considered statistically significant.
Results
Huperzine A Suppresses Glial and Endothelial Activation and Leukocyte Infiltration
Both activated glial cells, which are characterized by an increased level of the cell surface adhesion molecule ICAM-1, and activated endothelium cells, which are characterized by increased levels of E-selectin, VCAM-1 and ICAM-1, play critical roles in leukocyte recruitment during neurovascular inflammation. However, E-selectin mRNA levels were not significantly different among the 3 experimental groups (1-way ANOVA, F = 3.220, p = 0.112; table 2). We observed that D-gal significantly increased the mRNA levels of VCAM-1 and ICAM-1 expressed in hippocampal tissue when compared with those of the saline-treated control group (table 2). Huperzine A treatment reduced D-gal-induced VCAM-1 and ICAM-1 mRNA expression compared with the D-gal-treated group (table 2). There were significant differences in VCAM-1 and ICAM-1 mRNA expression levels between the D-gal + huperzine A group and the saline-treated control group.
mRNA levels of ICAM-1, VCAM-1 and E-selectin (means ± SEM)

The increased spatial distributions of GFAP, CD11b and E-selectin were observed in the CA1 and dentate gyrus areas of the D-gal-treated group (fig. 1a-c). Huperzine A significantly reduced the D-gal-induced number of activated astrocytes with hypertrophy and intensely stained processes (fig. 1a). Huperzine A also significantly attenuated the D-gal-induced increases in CD11b and E-selectin (fig. 1b, c). Furthermore, the increased protein levels of GFAP, E-selectin, VCAM-1, ICAM-1 and CD11b in the hippocampi of the D-gal-treated group were verified by Western blot analysis (fig. 2a-d). Huperzine A significantly but incompletely attenuated the D-gal-induced increases in these protein levels (fig. 2b, d). These data revealed that the AChEI huperzine A could significantly, but not completely, reduce the number of activated inflammatory cells, including glia and endothelial cells, as well as leukocyte recruitment, in the hippocampus.

Huperzine A blocks D-gal-induced glial and endothelial activation and leukocyte infiltration in the rat hippocampus. a The representative images were taken from the CA1 and dentate gyrus (DG) regions. Activated astrocytes with hypertrophy and intensely stained processes were observed in these regions in D-gal-treated rats. b Leukocyte infiltration with CD11b staining also increased in the CA1 and dentate gyrus regions in D-gal-treated rats. c Activated endothelial cells with intense E-selectin staining were seen in the CA1 and dentate gyrus regions in D-gal-treated rats.
Huperzine A blocks D-gal-induced glial and endothelial activation and leukocyte infiltration in the rat hippocampus. a The representative images were taken from the CA1 and dentate gyrus (DG) regions. Activated astrocytes with hypertrophy and intensely stained processes were observed in these regions in D-gal-treated rats. b Leukocyte infiltration with CD11b staining also increased in the CA1 and dentate gyrus regions in D-gal-treated rats. c Activated endothelial cells with intense E-selectin staining were seen in the CA1 and dentate gyrus regions in D-gal-treated rats.

a Expression of the adhesion molecules E-selectin, ICAM-1 and VCAM-1, leukocyte infiltration CD11b in rat hippocampi in different experimental conditions. Western blotting is the most representative out of 3 different consistent experiments. As shown, samples were normalized by incubating membranes in the presence of β-actin antibody. b Densitometric analysis performed on 3 different consistent experiments. The bands of these proteins were significantly increased in the D-gal group compared with both the saline-treated control group and D-gal + huperzine A group (n = 5). Significant changes in these protein levels were also observed between the saline-treated control group and D-gal + huperzine A group. c Expression of GFAP in rat hippocampi in different experimental conditions. Western blotting is the most representative out of 3 different consistent experiments. As shown, samples were normalized by incubating membranes in the presence of GAPDH antibody. d Densitometric analysis performed on 3 different consistent experiments. The protein bands were significantly increased in the D-gal group compared with both the saline-treated control group and D-gal + huperzine A group (n = 5). No significant changes between the saline-treated control group and D-gal + huperzine A group were observed. The data were presented as the mean ± SEM.
a Expression of the adhesion molecules E-selectin, ICAM-1 and VCAM-1, leukocyte infiltration CD11b in rat hippocampi in different experimental conditions. Western blotting is the most representative out of 3 different consistent experiments. As shown, samples were normalized by incubating membranes in the presence of β-actin antibody. b Densitometric analysis performed on 3 different consistent experiments. The bands of these proteins were significantly increased in the D-gal group compared with both the saline-treated control group and D-gal + huperzine A group (n = 5). Significant changes in these protein levels were also observed between the saline-treated control group and D-gal + huperzine A group. c Expression of GFAP in rat hippocampi in different experimental conditions. Western blotting is the most representative out of 3 different consistent experiments. As shown, samples were normalized by incubating membranes in the presence of GAPDH antibody. d Densitometric analysis performed on 3 different consistent experiments. The protein bands were significantly increased in the D-gal group compared with both the saline-treated control group and D-gal + huperzine A group (n = 5). No significant changes between the saline-treated control group and D-gal + huperzine A group were observed. The data were presented as the mean ± SEM.
Huperzine A Suppresses the D-Gal-Induced Release of Inflammatory Cytokines
Both glial and endothelial cell activation resulted in increased mRNA and protein levels of proinflammatory cytokines (table 3; fig. 3). The mRNA expression of the proinflammatory cytokines TNF-α, IL-1β and IL-6 was significantly increased in the D-gal-treated group compared with the saline-treated control group (table 3). The expression levels of the proinflammatory cytokines TNF-α, IL-1β and IL-6 mRNA in the D-gal + huperzine A group were significantly higher compared with the saline-treated control group and significantly lower compared with the D-gal group (table 3).
mRNA levels of TNF-α, IL-1β and IL-6 (means ± SEM)

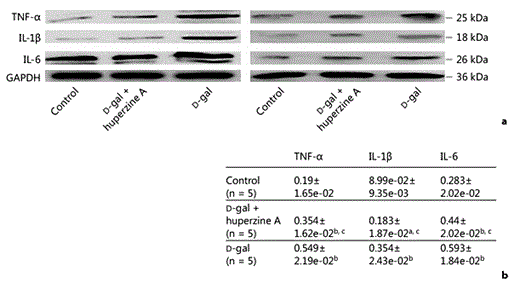
Huperzine A blocks the D-gal-induced increase in proinflammatory cytokines TNF-α, IL-1β and IL-6 in protein levels in the rat hippocampus. a Western blotting of 2 representative bands out of 3 different consistent experiments. As shown, samples were normalized by incubating membranes in the presence of GAPDH antibody. b Densitometric analysis performed on 3 different consistent experiments. These protein bands were significantly increased in the D-gal group compared with both the saline-treated control group and D-gal + huperzine A group (n = 5). Significant changes in the protein levels of TNF-α, IL-1β and IL-6 were also observed between the saline-treated control and D-gal + huperzine A groups. All of the data were presented as the mean ± SEM. a p < 0.01 and b p < 0.001 versus the saline-treated control group; c p < 0.001 versus the D-gal-treated group.
Huperzine A blocks the D-gal-induced increase in proinflammatory cytokines TNF-α, IL-1β and IL-6 in protein levels in the rat hippocampus. a Western blotting of 2 representative bands out of 3 different consistent experiments. As shown, samples were normalized by incubating membranes in the presence of GAPDH antibody. b Densitometric analysis performed on 3 different consistent experiments. These protein bands were significantly increased in the D-gal group compared with both the saline-treated control group and D-gal + huperzine A group (n = 5). Significant changes in the protein levels of TNF-α, IL-1β and IL-6 were also observed between the saline-treated control and D-gal + huperzine A groups. All of the data were presented as the mean ± SEM. a p < 0.01 and b p < 0.001 versus the saline-treated control group; c p < 0.001 versus the D-gal-treated group.
The protein expression of the proinflammatory cytokines TNF-α, IL-1β and IL-6 in the hippocampus exhibited a change similar to the mRNA levels in the 3 experimental groups. The TNF-α optical density value significantly increased in the D-gal group when compared with the saline-treated control and D-gal + huperzine A groups (fig. 3a, b). A similar change was also observed in the IL-1β and IL-6 optical density values of the D-gal group compared with the saline-treated control and D-gal + huperzine A groups (fig. 3a, b). The optical values for the D-gal + huperzine A group were similar to the mRNA levels and were thus higher than those of the saline-treated control group. There was a significant difference between the saline-treated control and D-gal + huperzine A groups in the levels of TNF-α, IL-1β and IL-6 (fig. 3a, b), in the hippocampal tissues.
Huperzine A Inhibits D-Gal-Induced Cell Apoptosis and Dysfunction of the BBB
RECA-1 is expressed on the cell surface of all rat endothelial cells [41, 42, 43, 44]. Immunostaining of both RECA-1 (red; colors refer to the online version only) and TUNEL (green) in CA2 and CA3 areas of representative rat hippocampal sections from the 3 experimental groups is shown in figure 4a. Apoptosis in the neurovascular units was significantly increased in the D-gal group when compared with the other 2 groups (fig. 4a). Apoptotic death in the neurovascular units in the hippocampus was further verified by measuring active caspase-3 levels. The level of active caspase-3 in the D-gal group was significantly increased compared with the saline-treated control and D-gal + huperzine A groups. Differences in the levels of active caspase-3 were not observed between the D-gal + huperzine A and the saline-treated control groups (fig. 4b). The decrease in endothelial cells in the hippocampus was verified by measurement of RECA-1 optical density (fig. 4b, c). There were significant differences in the levels of RECA-1 protein between the saline-treated control group and the D-gal group, between the D-gal group and D-gal + huperzine A group, and between the saline-treated control group and D-gal + huperzine A group from 3 independent experiments (fig. 4c, d). This result suggested that huperzine A significantly suppressed D-gal-induced apoptotic death in neurovascular units, including endothelial cells.

Huperzine A inhibits D-gal-induced cell apoptosis and prevents D-gal-induced decreases in the expression of the RECA-1 in the rat hippocampus. a The representative images showing the double staining of TUNEL (green; colors refer to the online version only) and RECA-1 antibody (red) in hippocampal sections in different experimental conditions were taken from the CA2 and CA3 regions. The yellow arrowheads indicate endothelial cell apoptosis. More apoptotic cell death (green) occurred in the neurovascular tissues in the D-gal group. The cellular nuclei were stained using 4′,6-diamidino-2-phenylindole (blue). b Expression of active caspase-3 and RECA-1 in rat hippocampi in different experimental conditions. Western blotting is the most representative out of 3 different consistent experiments. As shown, samples were normalized by incubating membranes in the presence of GAPDH antibody. c Densitometric analysis performed on 3 different consistent experiments. The active caspase-3 and RECA-1 band was significantly decreased in the D-gal group compared with both the saline-treated control group and D-gal + huperzine A group. All of the data were presented as the mean ± SEM. a p < 0.05 and b p < 0.001 versus the saline-treated control group; c p < 0.001 versus the D-gal-treated group.
Huperzine A inhibits D-gal-induced cell apoptosis and prevents D-gal-induced decreases in the expression of the RECA-1 in the rat hippocampus. a The representative images showing the double staining of TUNEL (green; colors refer to the online version only) and RECA-1 antibody (red) in hippocampal sections in different experimental conditions were taken from the CA2 and CA3 regions. The yellow arrowheads indicate endothelial cell apoptosis. More apoptotic cell death (green) occurred in the neurovascular tissues in the D-gal group. The cellular nuclei were stained using 4′,6-diamidino-2-phenylindole (blue). b Expression of active caspase-3 and RECA-1 in rat hippocampi in different experimental conditions. Western blotting is the most representative out of 3 different consistent experiments. As shown, samples were normalized by incubating membranes in the presence of GAPDH antibody. c Densitometric analysis performed on 3 different consistent experiments. The active caspase-3 and RECA-1 band was significantly decreased in the D-gal group compared with both the saline-treated control group and D-gal + huperzine A group. All of the data were presented as the mean ± SEM. a p < 0.05 and b p < 0.001 versus the saline-treated control group; c p < 0.001 versus the D-gal-treated group.
At the electron-microscopic level, the neurovascular units of the hippocampi of D-gal-treated rats exhibited clear morphological abnormalities (fig. 5a-c). Compared with the saline-treated control group and D-gal + huperzine A groups, swelling of the perisynaptic processes of astrocytes around the microvessels was evident in the D-gal group (fig. 5b, c). Senescence-like enlarged mitochondria with destroyed mitochondrial cristae were significantly increased in the D-gal group when compared with the D-gal + huperzine A and the saline-treated control groups (left column of fig. 5a-c). Close examination revealed that in the D-gal group, the TJs exhibited an abnormal appearance at the endothelial cell-cell contact regions, demonstrating that the density of the so-called kissing points of the TJs had significantly decreased (right column of fig. 5a-c).

Cerebrovascular ultrastructural analyses of the CA2 and CA3 regions in different experimental conditions. Ultrathin electron microscopy sections revealed the neurovascular units of the CA2 and CA3 regions in the saline-treated control group (a), in the D-gal + huperzine A group (b) and in the D-gal group (c). The right column shows the higher magnification of the left. D-gal-treated rats exhibited obvious morphological abnormalities. The swelling of the perisynaptic processes of astrocytes around the microvessels was evident in the D-gal group. The senescence-like enlarged mitochondria with damaged mitochondrial cristae were significantly increased in the D-gal group. The so-called kissing points of the TJs were clearly visualized at a higher magnification (white arrowheads). The optical density of the kissing points was significantly decreased in the D-gal group compared with both the saline-treated control group and D-gal + huperzine A group.
Cerebrovascular ultrastructural analyses of the CA2 and CA3 regions in different experimental conditions. Ultrathin electron microscopy sections revealed the neurovascular units of the CA2 and CA3 regions in the saline-treated control group (a), in the D-gal + huperzine A group (b) and in the D-gal group (c). The right column shows the higher magnification of the left. D-gal-treated rats exhibited obvious morphological abnormalities. The swelling of the perisynaptic processes of astrocytes around the microvessels was evident in the D-gal group. The senescence-like enlarged mitochondria with damaged mitochondrial cristae were significantly increased in the D-gal group. The so-called kissing points of the TJs were clearly visualized at a higher magnification (white arrowheads). The optical density of the kissing points was significantly decreased in the D-gal group compared with both the saline-treated control group and D-gal + huperzine A group.
As a marker of BBB disruption in the brain, the fluorescence intensity of IgG staining was significantly higher in the D-gal-treated group compared with the saline-treated control and D-gal + huperzine A groups. The fluorescence intensity of the IgG staining in the D-gal + huperzine A group was also significantly higher compared with the saline-treated control group (table 4).
IgG leakage (means ± SEM)

The proteins related to the transcellular and paracellular diffusion pathways are major determinants of BBB function (i.e. permeability). The transcellular pathway-related proteins consisted of occludin and Zo-1. The level of occludin mRNA relative to β-actin mRNA in the hippocampal tissue in the D-gal group was significantly altered compared with the saline-treated control group and the D-gal + huperzine A group (table 5). The Zo-1 mRNA level relative to β-actin mRNA in the hippocampal tissue in the D-gal group was significantly altered compared with the saline-treated control group and the D-gal and huperzine A group. There was no significant difference between the saline-treated control group and the D-gal + huperzine A group with respect to the relative occludin mRNA levels. However, the relative Zo-1 mRNA level for the D-gal + huperzine A group was significantly decreased compared with the saline-treated control group (table 5).
mRNA levels of occludin and Zo-1 (means ± SEM)

The density of the transcellular pathway-related proteins also paralleled the alterations of their corresponding mRNA levels. A representative Western blot of occludin and Zo-1 protein in the hippocampus is shown in figure 6a. The relative optical density values of occludin and Zo-1 protein in hippocampal tissues from the 3 experimental groups are summarized in figure 6b. Both the occludin and Zo-1 protein levels in hippocampal tissues of the D-gal group were significantly reduced compared with the saline-treated control group and D-gal + huperzine A group. There was a significant difference in the levels of occludin and Zo-1 between the saline-treated control and D-gal + huperzine A groups.

Huperzine A blocks D-gal-induced BBB dysfunction and the decrease in the BBB-related proteins occludin and Zo-1, and the TJ protein claudin-5 in the rat hippocampus. a Expression of the BBB-related proteins occludin and Zo-1 in rat hippocampi in different experimental conditions. Western blotting is the most representative out of 3 different consistent experiments. As shown, samples were normalized by incubating membranes in the presence of GAPDH antibody. b Densitometric analysis performed on 3 different consistent experiments. The two protein bands were significantly decreased in the D-gal group compared with both the saline-treated control group and D-gal + huperzine A group (n = 5). Significant changes in both the occludin and Zo-1 levels were observed between the saline-treated control group and D-gal + huperzine A group. c Expression of the TJ protein claudin-5 in rat hippocampi in different experimental conditions. Western blotting is the most representative out of 3 different consistent experiments. As shown, samples were normalized by incubating membranes in the presence of β-actin antibody. d Densitometric analysis performed on 3 different consistent experiments. The claudin-5 band was significantly decreased in the D-gal group compared with both the saline-treated control group and D-gal + huperzine A group (n = 3). Significant changes in claudin-5 level were also observed between the saline-treated control and D-gal + huperzine A groups. All of the data were presented as the mean ± SEM.
Huperzine A blocks D-gal-induced BBB dysfunction and the decrease in the BBB-related proteins occludin and Zo-1, and the TJ protein claudin-5 in the rat hippocampus. a Expression of the BBB-related proteins occludin and Zo-1 in rat hippocampi in different experimental conditions. Western blotting is the most representative out of 3 different consistent experiments. As shown, samples were normalized by incubating membranes in the presence of GAPDH antibody. b Densitometric analysis performed on 3 different consistent experiments. The two protein bands were significantly decreased in the D-gal group compared with both the saline-treated control group and D-gal + huperzine A group (n = 5). Significant changes in both the occludin and Zo-1 levels were observed between the saline-treated control group and D-gal + huperzine A group. c Expression of the TJ protein claudin-5 in rat hippocampi in different experimental conditions. Western blotting is the most representative out of 3 different consistent experiments. As shown, samples were normalized by incubating membranes in the presence of β-actin antibody. d Densitometric analysis performed on 3 different consistent experiments. The claudin-5 band was significantly decreased in the D-gal group compared with both the saline-treated control group and D-gal + huperzine A group (n = 3). Significant changes in claudin-5 level were also observed between the saline-treated control and D-gal + huperzine A groups. All of the data were presented as the mean ± SEM.
Claudin-5, an endothelial TJ density protein, plays an important role in the size-selective relaxation of the BBB [23]. The relative optical density values of the claudin-5 protein were measured in hippocampal tissues from 3 independent experiments. The level of claudin-5 in hippocampal tissues of the D-gal group was significantly reduced compared with the saline-treated control group and D-gal + huperzine A group (fig. 6c, d). Huperzine A did not completely restore the D-gal-induced decrease in claudin-5 protein. The difference in the claudin-5 protein level between the saline-treated control group and the D-gal + huperzine A group was also evident (fig. 6c, d).
Huperzine A Suppresses Neurovascular Damage by Inhibiting IκBα Degradation and NF-κB Nuclear Localization
Nuclear p65 expression was significantly increased in the D-gal group compared with the saline-treated control group and D-gal + huperzine A group (fig. 7a, b). Huperzine A failed to completely suppress D-gal-induced p65 nuclear translocation. However, there was a significant difference between the saline-treated control group and D-gal + huperzine A group (fig. 7a, b). Nuclear phospho-p65 protein was only detected in the D-gal group (fig. 7a). Thus, huperzine A significantly inhibited D-gal-induced NF-κB activity.

Huperzine A suppressed D-gal-induced p65 nuclear translocation and activation. a Expression of phosphorylated p65 and nuclear protein p65 in rat hippocampi in different experimental conditions. Western blotting is the most representative out of 3 different consistent experiments. As shown, samples were normalized by incubating membranes in the presence of His antibody. A weak band of phosphorylated p65 was observed in the D-gal group. There was no phosphorylated p65 band in both the saline-treated control group and D-gal + huperzine A group. b Densitometric analysis performed on 3 different consistent experiments. The p65 band was significantly increased in the D-gal group compared with both the saline-treated control group and D-gal + huperzine A group (n = 5). Significant changes in the p65 levels were also observed between the saline-treated control and D-gal + huperzine A groups. a p < 0.001 versus the saline-treated control group; b p < 0.001 versus the D-gal-treated group. c Huperzine A suppressed D-gal-induced IκBα degradation and cytoplasmic p65 nuclear translocation in the rat hippocampus. Western blotting is the most representative out of 3 different consistent experiments. As shown, samples were normalized by incubating membranes in the presence of GAPDH antibody. d Densitometric analysis performed on 3 different consistent experiments. Both the IκBα and p65 bands were significantly decreased in the D-gal group compared with both the saline-treated control group and D-gal + huperzine A group (n = 5). No significant changes in both the IκBα and p65 level were observed between the saline-treated control and D-gal + huperzine A groups. All of the data were presented as the mean ± SEM. a p < 0.001 versus the saline-treated control group; b p < 0.001 versus the D-gal-treated group.
Huperzine A suppressed D-gal-induced p65 nuclear translocation and activation. a Expression of phosphorylated p65 and nuclear protein p65 in rat hippocampi in different experimental conditions. Western blotting is the most representative out of 3 different consistent experiments. As shown, samples were normalized by incubating membranes in the presence of His antibody. A weak band of phosphorylated p65 was observed in the D-gal group. There was no phosphorylated p65 band in both the saline-treated control group and D-gal + huperzine A group. b Densitometric analysis performed on 3 different consistent experiments. The p65 band was significantly increased in the D-gal group compared with both the saline-treated control group and D-gal + huperzine A group (n = 5). Significant changes in the p65 levels were also observed between the saline-treated control and D-gal + huperzine A groups. a p < 0.001 versus the saline-treated control group; b p < 0.001 versus the D-gal-treated group. c Huperzine A suppressed D-gal-induced IκBα degradation and cytoplasmic p65 nuclear translocation in the rat hippocampus. Western blotting is the most representative out of 3 different consistent experiments. As shown, samples were normalized by incubating membranes in the presence of GAPDH antibody. d Densitometric analysis performed on 3 different consistent experiments. Both the IκBα and p65 bands were significantly decreased in the D-gal group compared with both the saline-treated control group and D-gal + huperzine A group (n = 5). No significant changes in both the IκBα and p65 level were observed between the saline-treated control and D-gal + huperzine A groups. All of the data were presented as the mean ± SEM. a p < 0.001 versus the saline-treated control group; b p < 0.001 versus the D-gal-treated group.
In the rat hippocampus, there was a significant low level of cytoplasmic IκBα due to degradation in the D-gal group compared with the saline-treated control group and D-gal + huperzine A group (fig. 7c, d). However, the difference in the IκBα level between the D-gal + huperzine A and the saline-treated control groups was not significant. The cytoplasmic p65 levels exhibited significant changes among the 3 experimental groups (F = 503.152, p < 0.001, 1-way ANOVA). The cytoplasmic p65 level was lowest in the D-gal group when compared with the saline-treated control group and D-gal + huperzine A group. Additionally, there was a significant difference in the cytoplasmic p65 level between the saline-treated control group and D-gal + huperzine A group (fig. 7c, d).
Huperzine A Might Slow the Inflammatory Responses by Inhibiting AChE Activity
AChE activity was significantly increased in the D-gal-treated group, and huperzine A treatment significantly suppressed the D-gal-induced increase in AChE activity in rat cerebral cortices (F = 47.434, p < 0.001, 1-way ANOVA; table 6) and hippocampi (F = 109.783, p < 0.001, 1-way ANOVA; table 6) from 3 independent experiments. However, the AChE activity in the D-gal + huperzine A group remained significantly lower than that of the saline-treated control group in hippocampi, but not in cortices (table 6).
Huperzine A inhibited the AChE activity in D-gal-treated rat cerebral cortex and hippocampus (means ± SEM)

Discussion
This study demonstrated that D-gal induced the activation of glial and vascular endothelial cells, infiltration of CD11b+ cells, release of proinflammatory cytokines, inflammatory damage in neurovascular units and a decrease in BBB-related proteins, which were associated with IκBα degradation and nuclear translocation of NF-κB in the rat hippocampus. CD11b is predominantly expressed in monocytes and granulocytes, also expressed in microglia. CD11b belongs to the integrin α-chain family and is implicated in various adhesive interactions of monocytes, macrophages and granulocytes as well as in mediating the uptake of complement-coated particles. Huperzine A reduced D-gal-induced neurovascular inflammation, improved BBB function, increased the expression of the BBB-related proteins occludin, Zo-1 and the TJ protein claudin-5, and suppressed apoptosis in the rat hippocampus. Huperzine A also blocked D-gal-induced chronic sterile inflammation by inhibiting cytoplasmic IκBα degradation and NF-κB activation. Thus, this anti-inflammatory function might be an important mechanism of neurovascular protection via huperzine A. The inhibition by huperzine A of AChE activity might be associated with its neurovascular protective effects.
Inflammation in the absence of any microorganism has been termed sterile inflammation. Recent studies have demonstrated that DAMPs and intercellular cytokines produced due to mitochondrial DNA damage and oxidative stress were required for NLRP3/NALP3 inflammasome activation. Activated inflammasomes can direct the proteolytic maturation of inflammatory cytokines such as IL-1β [12, 45]. Accumulated DAMPs, including AGEs arising from nonenzymatic glycation and oxidation of proteins and lipids, may also be detected by non-PRR-mediated recognition [12]. The prototypical DAMP-specific receptor for AGEs was also found in endothelial cells [46]. D-gal not only causes mitochondrial dysfunction but also reacts with free amino acids and peptides to form AGEs. These events resulted in D-gal-induced sterile inflammation and cellular damage [6, 7, 11]. On the one hand, inflammation was beneficial for the repair of damaged tissue and the eradication of harmful pathogens. On the other hand, chronic inflammation was detrimental to the host when failure to remove the offending agent occurs. Sterile inflammation-related diseases will occur due to sustained production of DAMPs, ROS, proteases and growth factors by inflammatory cells [12]. The major targets of oxidative damage were lipids, proteins and DNA [47]. Low levels of ROS are generally required for normal physiological functions, such as cell growth, stress adaptation and injury response. In contrast, unregulated high levels of ROS are linked to pathological processes, such as cellular function impairment, ongoing tissue injury, and cellular apoptosis [48]. In another study, we found that mitochondrial DNA damage and ROS production played important roles in D-gal-induced senescent hepatic cells [10]. As expected, D-gal induced inflammatory injury, including apoptosis, BBB dysfunction and BBB-related protein depletion (fig. 4, 5, 6).
Neurovascular inflammation induced by ROS has been shown to be associated with BBB damage [49, 50]. Vascular endothelial cells are more vulnerable to oxidative stress than other nonneural cell types in the neurovascular unit, including astrocytes, microglia and pericytes, which are in close contact with endothelial cells [47, 51]. A previous study found that impaired function with aging in vascular endothelial cells of healthy humans was associated with the development of a proinflammatory phenotype in the vascular endothelium [49]. In our D-gal-induced senescent rat model, D-gal-induced chronic sterile inflammation in endothelial cells appeared to play an important role in the depletion of TJ-related proteins measured in the rat hippocampus. We demonstrated significant endothelial cell activation and a proinflammatory phenotype in the cerebrovascular endothelium of the hippocampus. The mRNA and/or protein levels of the endothelial cell-specific adhesion molecule E-selectin, other endothelial and glial cell-associated adhesion molecules, including ICAM-1 and VCAM-1, and proinflammatory cytokines, such as TNF-α, IL-1β and IL-6, were significantly increased in the hippocampus (fig. 1, 2, 3). This sterile inflammation in endothelial cells involved the NF-κB inflammatory signaling pathway, including IκBα degradation and NF-κB nuclear translocation and activation [49]. In the present study, cytoplasmic IκBα degradation and NF-κB nuclear translocation might also be associated with D-gal-induced endothelial cell activation and an inflammatory phenotype in the cerebrovascular endothelium of the hippocampus (fig. 1, 2, 7).
Glial activation and neuroinflammation contributed to the damage of BBB components by activating Toll-like receptor 4 following ischemic brain injury. Toll-like receptor 4 mediates several downstream pathways, such as the NF-κB, JAK-STAT and JNK/SAPK pathways [50]. Activated astroglia and microglia release proinflammatory cytokines, including TNF-α and IL-1β, via the NF-κB pathway [19]. The inflammatory activation of glia and the occurrence of senescence-like enlarged mitochondria with destroyed mitochondrial cristae were significantly increased in the D-gal group (fig. 1a, 2c, d, 5b, c). Thus, glial activation likely contributed to the increase in proinflammatory cytokines and the degradation of IκBα in the cytoplasm in the neurovascular unit (fig. 3, 7).
One important anatomical structure in the cerebrovascular endothelium is the endothelial TJ. Together with integral membrane proteins, such as occludin, and cytoplasmic proteins in endothelial cells, such as Zo-1, TJs create a rate-limiting barrier (the BBB). Aging rats exhibited significant alterations in BBB-related protein expression and BBB function [26, 27]. Decreases in BBB-related proteins are associated with an inflammatory response, vascular injury and apoptosis [47, 49, 52]. In the subgranular zone of 12-month-old male Fischer 344 rats, there was a significant decrease in the overall capillary volume [44]. In the present study, at the electron-microscopic level, there was a clear decrease in the density of TJs in the microvessels of the D-gal-induced hippocampus (fig. 5a-c). The increased IgG extravasation in the brain sections of the D-gal-treated group demonstrated significant BBB impairment (table 4). Thus, BBB dysfunction and a decrease in D-gal-induced BBB-related proteins might be caused by inflammatory damage of endothelial cells. The RECA-1 optical density value in the D-gal group was significantly decreased compared with the other two groups and was the highest in the saline-treated control group (fig. 4b, c). We speculated that huperzine A not only suppressed endothelial cell apoptosis but also inhibited endothelial cell senescence by increasing endothelial cell proliferation. Taken together, this hypothesis warrants further investigation in the future.
Cholinergic stimulation modulates host inflammatory responses via the cholinergic anti-inflammatory pathway [53, 54, 55]. The neurotransmitter acetylcholine (ACh), as well as nicotinic acetylcholine receptor (nAChR) agonists, blocks lipopolysaccharide-induced TNF release from macrophages via the α7-nAChR [53, 56]. Cholinergic nerve stimulation attenuates systemic inflammatory responses via the α7-nAChR-mediated anti-inflammatory pathway [53, 54, 56]. Moreover, a nonneuronal cholinergic system has been found in endothelial, glial and immune cells [18, 55, 57]. During inflammation, cholinergic stimulation blocks endothelial cell activation and leukocyte recruitment in vivo by inhibiting IκBα degradation and NF-κB nuclear translocation. Activation of the cholinergic anti-inflammatory pathway via the α7-nAChR plays a critical role in this process [18]. Vascular endothelial cells express the α7-nAChR, and the cholinergic agonist nicotine significantly reduces endothelial cell activation and modulates cerebral microvascular permeability [18]. AChEIs, including edrophonium, pyridostigmine and huperzine A, can inhibit the inflammatory response in lymphocytes or microglia by increasing the extracellular ACh concentration, which activates the α7-nAChR [19, 58]. Due to the short half-life and toxicity of nAChR agonists, such as nicotine and ACh, their use as therapeutic agents is limited. Huperzine A, a reversible and selective AChEI, is an effective intervention drug used to suppress both innate and adaptive inflammatory responses to ischemic brain injury, Alzheimer's disease and myasthenia gravis [19, 32, 34, 58]. In another study, we found that huperzine A also significantly suppressed D-gal-induced hepatic cell replicative senescence by inhibiting NF-κB-mediated inflammation [10]. One specific biomarker used to characterize the chronic administration of D-gal-induced aging in mice was the marked increase in AChE [6]. Despite the higher dose of D-gal that was administered to the rats, the present study indicated that AChE activity was significantly increased in the D-gal-treated group, which was consistent with a previous report in mice [6]. Huperzine A could significantly inhibit AChE activity of D-gal-treated rats (table 6). Furthermore, AChE activity in the D-gal + huperzine A group was significantly lower than that of the saline-treated control group in hippocampi (table 6). Therefore, it would be interesting to determine whether the pharmacological profiles of huperzine A described above are associated with its potent AChE inhibitory effect. A previous study demonstrated that huperzine A exhibits cholinergic anti-inflammatory and neuroprotective effects in a rat model of transient focal cerebral ischemia via activation of the nAChR [19]. In another pilot study, we found that the nAChR antagonist mecamylamine can abolish the inhibitory effects of huperzine A on glial activation and neuroinflammation of naturally aged C57 mice. During the early stage of D-gal-induced neuroinflammation, activated astrocytes in the mouse hippocampus exhibited upregulated expression of glutamate transporter 1 and aquaporin 4, which might result in decreased glutamate in the synaptic cleft and reduced glutamate excitotoxicity by increasing its uptake by astrocytes [59]. However, L-glutamate released from activated microglia downregulated astrocytic L-glutamate transporter expression and resulted in the elevation of extracellular L-glutamate [60]. Huperzine A could block the N-methyl-D-aspartate receptors and protect against diverse neurodegenerative diseases [37]. Therefore, further studies are required to understand the mechanism of the protective effects of huperzine A on D-gal-induced inflammatory impairment of neurovascular tissues.
Acknowledgments
This work was supported by the Shanghai Key Laboratory of Clinical Geriatric Medicine Subject Construction (No. 13dz2260700) and Grants-in-Aid for Scientific Research from the Shanghai Municipal Health Bureau (No. 2011-4144 and XBR 2013091).
Disclosure Statement
The authors declare that they have no competing interests to disclose.
References
Additional information
Q.R., X.H. and H.A. contributed equally to this work.
