

Abstract
Cell senescence is one of the major paradigms of aging research. It started with the demonstration by L. Hayflick of the limited number of divisions by normal, nontransformed cells, not shown by transformed malignant cells, this processes being largely regulated by the telomere-telomerase system. A complete renewal of this discipline came from the demonstration that cells can enter senescence at any time by an anti-oncogene-triggered pathway, enabling them to escape malignancy. The senescent cell became a major actor of the aging process, among others, by the acquisition of the senescence-associated secretory phenotype. This chapter is devoted to the regulatory process involved in the acquisition of the senescent cell phenotype and its role in organismal aging.
To understand aging, as felt by persons and studied by medical science through age-related diseases, is an ancient preoccupation of humanity and research. In order to understand its multiple facets, the traditional ‘method' of experimental medicine, model experiments, is of crucial importance. This was largely facilitated when cell and tissue culture methods were developed. Current methods for cell cultures were developed only during the second part of the 20th century, with the exception of some earlier attempts we shall shortly mention. Early studies were carried out by Alexis Carrel, a French surgeon from Lyon who worked during most of his carrier at the Rockefeller Institute in New York City. He used explant cultures of chicken embryos, maintained in a nutritional medium containing blood plasma and chick embryo extracts, obtained by grinding young chick embryos and using the filtrate of this tissue suspension. He showed that chick embryo heart explants could continue to beat, and muscle explants could contract upon stimulation, for a long time.
Interested in aging, Carrel noticed that such explant cultures could be kept in sterile conditions at 37°C for much longer than the life expectancy of the animal. He concluded that disease and death are mainly due to body fluids, ‘humors' which would progressively alter with age and harm the organism. Carrel was successfully contradicted in the 1960s when Leonard Hayflick made his important observations on cell cultures and showed that normal, nontransformed cells cannot divide indefinitely. Hayflick's findings were not immediately accepted. He investigated closely Carrel's methodology and even interviewed Carrel's former technician, retired in the ‘colonies'. She told Hayflick that the embryo extract had to be prepared in Lyon and delivered by airplane to Carrel's lab in New York. As it was filtered during its preparation using a tissue (gauze), it could contain fresh living cells this could explain the apparently unlimited survival of the cultures. Other scientists who attempted to reproduce Carrel's findings, invariably failed, and Hayflick's paradigm was progressively accepted, not however without repeated denials and criticisms. Most scientists who worked according to the guidelines published by Hayflick could reproduce his results. During the 2nd part of the last century, cell and tissue culture methods became standardized, and culture dishes and media became commercially available. This largely contributed to the interlaboratory standardization of culture methodology and settled to a large extent the controversies. Hayflick's paradigm, stating that normal nontransformed cells cannot duplicate indefinitely in culture unless transformed into malignant cells, is now largely accepted.
Cell Aging in vitro: The Hayflick Paradigm
Hayflick's methodology, developed with great care and precision, established a highly standardized culture procedure which yielded important results. Among them, for the purpose of this chapter, were that normal human cells in culture carry out a limited number of divisions (doubling of cells propagated with a 1:2 split ratio), as described in the original publication [1] as well as in reviews [2,3]. A second important experiment, repeated by several laboratories showed that the maximal number of cell divisions reached by normal human cell cultures decreased with the age of the donor (fig. 1) [for review see [4]]. However, there is considerable interindividual variation, and so the notion that cells from older individuals complete fewer population doublings than cells from younger individuals is not a fixed rule.
![Age of the donor vs. average population doublings of the respective skin fibroblasts derived from a human embryo (964 S) and from patients with benign tumors (⚫) or breast cancers (⚪). The straight line is the regression fitting the plot corresponding to the fibroblasts originating from donors with benign lesions. The correlation coefficient is indicated on the chart. Reproduced with permission from Macieira-Coelho [4].](https://karger.silverchair-cdn.com/karger/content_public/books/210/parts/chapter_358899/2/m_154042.jpeg?Expires=1716297056&Signature=V4Y4S2Kz6e4-cpjxF3kFT3ZMpNeFvpi-mRurmy065z~xwIjiBt-20tzQTwQ3GBatMRD-pIQAA84bAaie2ZcKUJDYwxB40BPkutFeI1P8cwRBd9UiSuEYXr~C8Yo1E6YZTurMHdR3UUlzdEcSnzIrMpv6aUf1rMGjLP4k5dtekbLcjl1QHpeQCE0RRhaZSDa7owsMCE6Mnfo3UhKn-kFFh7FifYObWPWKGxynxAVQuu7yODtmVa-49CAVGN~wSY8SnNJE~mfG78WTMR6G-hk3~H5OA5xZdDdLJyktN0iHIijRT5tAPLiPM6Q030l6HJR4q74249KlQqc2kk5Tnt49Iw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Age of the donor vs. average population doublings of the respective skin fibroblasts derived from a human embryo (964 S) and from patients with benign tumors (⚫) or breast cancers (⚪). The straight line is the regression fitting the plot corresponding to the fibroblasts originating from donors with benign lesions. The correlation coefficient is indicated on the chart. Reproduced with permission from Macieira-Coelho [4].
Age of the donor vs. average population doublings of the respective skin fibroblasts derived from a human embryo (964 S) and from patients with benign tumors (⚫) or breast cancers (⚪). The straight line is the regression fitting the plot corresponding to the fibroblasts originating from donors with benign lesions. The correlation coefficient is indicated on the chart. Reproduced with permission from Macieira-Coelho [4].
A third observation was the occasional spontaneous transformation of cultured rodent (mice, rats) cells from the normal state of finite cell division capacity to a premalignant or malignant state of unlimited cell divisions [4]. This was shown more recently to be partly attributable to the expression of telomerase by most malignant cells [5]. There would be much more to say about the work of Hayflick and his followers. For this chapter, the most important conclusions are those previously stated: the limited number of population doublings by normal, nontransformed cell cultures, the general decrease with age of the maximal cell proliferation capacity and the loss of these properties after malignant transformation. For more information, the chapters by Robin Holliday and Alvaro Macieira-Coelho can be consulted.
End of Proliferation: The Senescent Phenotype
Those teams engaged in the above research mostly agreed with Hayflick's proposition, summarized in figure 2. It shows the successive phases of a normal (human) embryonic fibroblast cell culture, as described by Hayflick. After an initial slow proliferation, there followed a phase of near-linear growth, which ended in a decline of proliferative capacity during which cells enlarged, became vacuolated and were unable to enter the cell cycle (fig. 3). Not much interest arose about the fate of those cells, although it was acknowledged that they were not dead. A curious anecdote comes to my mind (L.R.) in this respect which clearly shows the confusion concerning this end stage of cell cultures. A well-known international publisher asked me to read a book published in the US by a scientist on the west coast to decide if it should be translated and published in France. To my astonishment and to make a long story short, I read in these pages that phase III (senescent) cells as designated by Hayflick are dead! I did not recommend the translation of this book into French.
![Diagrammatic representation of the history of cell strains and the phenomenon of cell alteration. In phase I, the primary culture terminates with the formation of the first confluent sheet. Phase II is characterized by luxuriant growth necessitating many subcultivations. Cells in this phase are termed ‘cell strains'. An alteration may occur at any time, giving rise to a ‘cell line' whose potential life is infinite. Conversely, cell strains characteristically enter phase III and are lost after a finite period of time. Reproduced with permission from Hayflick [3].](https://karger.silverchair-cdn.com/karger/content_public/books/210/parts/chapter_358899/2/m_154041.jpeg?Expires=1716297056&Signature=DYhktBqS9ogx49o-eBRBt46QDsh3pYrOMMrWsTg8VHn1rj6MeJkIE4sGUELsQpglU2iOgN7PsLN5HWjkPzdHqbjfTrNWCpVGKkt9VOh9jGVCo08ZK3tWolF~Y3egi93iyf1TJktocx9OOJ-GwZa2PBvLAQQGveK~DzWw7Jk21o7BBJ-sW~ObT3vOf-TfGxw3L3BBPt1GaWF1Lc3sadXZsmECIwxyPRDk~NGTXNITqvjMOXal2edvNOufDj~huKGhlc-OiQWZWyI8pAbLd8GmQo995NOdQ4z9WgKS0TiQvOPaVKrX~XMjB1wI1jrRZGY9lxXzaOIyDMNHhpVfB~BgxQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Diagrammatic representation of the history of cell strains and the phenomenon of cell alteration. In phase I, the primary culture terminates with the formation of the first confluent sheet. Phase II is characterized by luxuriant growth necessitating many subcultivations. Cells in this phase are termed ‘cell strains'. An alteration may occur at any time, giving rise to a ‘cell line' whose potential life is infinite. Conversely, cell strains characteristically enter phase III and are lost after a finite period of time. Reproduced with permission from Hayflick [3].
Diagrammatic representation of the history of cell strains and the phenomenon of cell alteration. In phase I, the primary culture terminates with the formation of the first confluent sheet. Phase II is characterized by luxuriant growth necessitating many subcultivations. Cells in this phase are termed ‘cell strains'. An alteration may occur at any time, giving rise to a ‘cell line' whose potential life is infinite. Conversely, cell strains characteristically enter phase III and are lost after a finite period of time. Reproduced with permission from Hayflick [3].
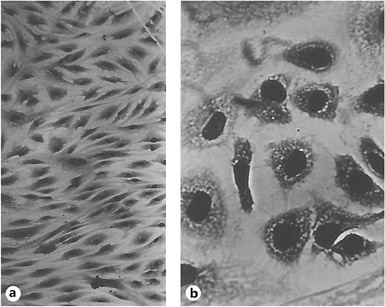
Microscopic picture of early passage (‘young') fibroblasts (a) and fibroblasts at the end of their replicative potential (b). Photos by L. Hayflick, reproduced with permission.
Microscopic picture of early passage (‘young') fibroblasts (a) and fibroblasts at the end of their replicative potential (b). Photos by L. Hayflick, reproduced with permission.
Here ends - more or less - the compressed story on this first, quite heroic era of the study of cellular senescence. One of its important results was acceptance of a culture model to study certain aspects of aging. Among the information that arose from these early studies were results concerning the fate of the genome during repeated cell division. Two of the most prolific and competent scientists, Drs. Robin Holliday and Alvaro Macieira-Coelho contribute chapters on this subject in this volume.
The Senescent Cell
Renewed interest in cellular senescence coincided with studies initiated by one of us (J.C.) Among the early results was the possibility of entering the senescent state without repeated proliferation, as shown by Hayflick. This possibility was experimentally confirmed and provided new insights into cellular senescence, as will be explained. Early on, it was suggested that cells might enter a senescent state in order to prevent malignant transformation, and thus cancer [6,7]. This idea attracted wide interest as shown by the large number of relevant publications, confirming the idea and attracting as well cancer biologists [for review see [8]]. It was proposed that entry into the senescent state was mediated by - at least - two different pathways linked to the activation of two tumor suppressive pathways, the p53/p21 and p16INK4a/pRB pathways (fig. 4). Both pathways are complex, having several upstream regulators and downstream effectors as well as modifying side branches, and ‘cross talk' between the pathways. One difference between the pathways is the p16INK4a/pRB pathway which imposes an essentially irreversible arrest of cell proliferation, whereas p53/p21 does not necessarily result in an irreversible growth arrest. p53 and pRB are master transcriptional regulators, p21 is a downstream effector of p53, and p16INK4a is a positive upstream regulator of pRB. p21 and p16INK4a are cyclin-dependent kinase inhibitors and, as such, potent inhibitors of the cell cycle. Other pathways that establish the senescent state may also exist, but details regarding such pathways are scant.
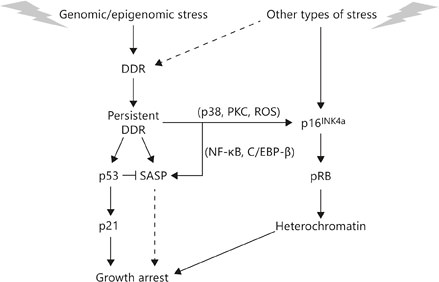
Regulation of senescence growth arrest and SASP. Cellular senescence is initiated by genomic or epigenomic damage, which activates a DDR. The DDR ultimately becomes persistent or chronic, which leads to activation of p38MAPK and protein kinase C (PKC) and increased ROS, and ultimately expression of the p16INK4a tumor suppressor. Stress that does not entail direct genomic or epigenomic damage can also induce p16INK4a expression, and in some cases can indirectly trigger a DDR (dashed line). p16INK4a activates the pBR tumor suppressor, which silences certain proproliferative genes by heterochromatinization, thereby instituting a stringent arrest of cell proliferation. Persistent DDR signaling also induces SASP and activates the p53 tumor suppressor, which restrains the SASP. p53 also causes growth arrest, principally by inducing expression of the cell cycle inhibitor p21. In some forms of oncogene-induced senescence, the SASP reinforces the senescence growth arrest (dashed line).
Regulation of senescence growth arrest and SASP. Cellular senescence is initiated by genomic or epigenomic damage, which activates a DDR. The DDR ultimately becomes persistent or chronic, which leads to activation of p38MAPK and protein kinase C (PKC) and increased ROS, and ultimately expression of the p16INK4a tumor suppressor. Stress that does not entail direct genomic or epigenomic damage can also induce p16INK4a expression, and in some cases can indirectly trigger a DDR (dashed line). p16INK4a activates the pBR tumor suppressor, which silences certain proproliferative genes by heterochromatinization, thereby instituting a stringent arrest of cell proliferation. Persistent DDR signaling also induces SASP and activates the p53 tumor suppressor, which restrains the SASP. p53 also causes growth arrest, principally by inducing expression of the cell cycle inhibitor p21. In some forms of oncogene-induced senescence, the SASP reinforces the senescence growth arrest (dashed line).
Among the stimuli that induce a senescent response is genomic damage, including telomere dysfunction. Genomic damage activates the DNA damage response (DDR) which in turn activates the p53/p21 pathway. The initial response to genomic damage is a rapid and robust DDR, within minutes or maximally hours, which is transient. If the damage is severe enough to elicit a senescence response, low-level DDR signaling, including activation of the p53/p21 pathway [9] may persist. Persistent DDR-signaling maintains the senescence growth arrest and might activate other signaling pathways such as those controlled by the stress-activated kinases p38MAPK and PKC. Senescent cells also contain elevated levels of reactive oxygen species (ROS), which can trigger other signaling pathways. Some of these ROS-initiated pathways will stimulate p16INK4a/pRB, the pathway responsible for the irreversibility of growth arrest [10]. The senescent state of such growth-arrested cells was studied and defined by several teams [11].
Senescent cells can be characterized by at least two procedures; the first and most used method [12] consists in staining cell cultures or tissue sections by a specific procedure visualizing cells expressing a pH6-optimal β-galactosidase, designated senescence associated or SA-β-Gal, shown to be a reliable marker of senescent cells. This method was used to show that senescent cells do indeed accumulate with age in tissues, in vivo. Similar results were obtained in sequential cell cultures too, carried out according to the Hayflick procedure (fig. 5). Using human skin fibroblasts, it could be shown that SA-β-Gal-positive cells accumulate during sequential passages [13]. It was then tested if advanced glycation end products (AGEs) of the Maillard reaction, known to play an important role in age-dependent modifications of tissues, as collagen crosslinking [14] could increase SA-β-Gal accumulation. As shown on table 1, this was observed indeed when AGEs, prepared as described were added to the fibroblast cultures [13]. A number of authors used this procedure for studies on cell aging all over the world [11].
Effect of an AGE on cell senescence in cultures quantified by SA-β-Gal expression in human dermal fibroblasts at different passages

![Increase with time in culture (4 and 7 days) and with increasing passages (abscissa) of the expression of SA-β-Gal, a marker of senescent cells in human skin fibroblasts at sequential passages, determined by the SA-β-Gal procedure [12]. Sequential cultures were started after 4 days in culture (lower curve) or after 7 days in culture (upper curve). Reproduced with permission [3].](https://karger.silverchair-cdn.com/karger/content_public/books/210/parts/chapter_358899/2/m_154038.jpeg?Expires=1716297056&Signature=XXUW-MsaHFoqKe5g4Dp6bssfxhATjFrVBFrqomw665H-DzBO9gdoWqDN-2ODq2p4kc6t6Z8IC9jLUZGKhFB4QKIGrmgTEmU6QRqcJrvp-58cZKbFUhQvbZ~iCugbpXCB6AAtbHU-GykQ9P8nTHN9QbloKg4RLnKKzD~FTQir9HwxPHeKcCVYSUSzgfLbn5VXITekKEHBEqc1qzeTrYPSPIM8fcXIJbtX-4frM56CYm-moytTtF2VbHDrx-5PoRm7LYrHBgs351LgPFaGfP~cQh0IeZ2lyTWmFJOofgEE4WqzcIFYc~Co90V4EW-7fmUNJQ7QVxc~nuOSRCNUBGoQCA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Increase with time in culture (4 and 7 days) and with increasing passages (abscissa) of the expression of SA-β-Gal, a marker of senescent cells in human skin fibroblasts at sequential passages, determined by the SA-β-Gal procedure [12]. Sequential cultures were started after 4 days in culture (lower curve) or after 7 days in culture (upper curve). Reproduced with permission [3].
Increase with time in culture (4 and 7 days) and with increasing passages (abscissa) of the expression of SA-β-Gal, a marker of senescent cells in human skin fibroblasts at sequential passages, determined by the SA-β-Gal procedure [12]. Sequential cultures were started after 4 days in culture (lower curve) or after 7 days in culture (upper curve). Reproduced with permission [3].
Another marker used for tracing cells entering senescence is the expression of the tumor suppressor gene p16INK4a. Its expression is low or undetectable in most normal, untransformed cells and tissues, but becomes readily detectable after induction of senescence by a variety of stimuli [15,16,17]. As for SA-β-Gal, the expression of this tumor suppressor gene was also shown to increase with age in several vertebrate tissues [18,19]. Most senescence inducers were shown to cause genomic damage, as shown by lasting DNA damage loci and DNA damage signaling (DDR). Such loci are designated by the acronym telomere dysfunction-induced loci or TIFs [20] when located at telomere regions, or DNA-SCARS (DNA segments with chromatin alterations reinforcing senescence [21]). Such loci exhibit several markers of DNA damage such as 53BP1, although distinct from loci formed immediately after DNA damage. DNA-SCARS can colocalize with PML (promyelocytic leukemia protein), nuclear bodies, exhibiting also activated DDR proteins as phospho-CHK2 directly involved in the senescence-associated secretory phenotype (SASP) response [22]. Persistent DNA damage loci were detected in tissues suffering genotoxic stress [22], as well as in aging mouse and primate tissues [20,23,24]. Senescence-associated heterochromatin loci were seen in some senescent cells, associated with silencing of some proproliferative genes [25,26]. Upregulation of the tumor suppressor protein DECI (deleted in esophageal cancer) and also DcR2 (decay receptor 2) [27] are also observed, both behaving as targets for p53 transactivation. Senescent cells also downregulate the expression of the nuclear lamina protein, lamin B1 [28,29]. Several other markers are less frequently used and are not detailed here.
The Senescence-Associated Secretory Phenotype
This feature of the senescent phenotype is expressed by a large number of senescent cells, considered as their most striking characteristic and probably involved in the development of organismal senescence as well as in age-related diseases [30,31] (fig. 6). Its development involves a large number of cytokines, chemokines, growth factors and proteases [31,32]. According to the nature of factors involved in the activation of the secretory phenotype, senescent cells can participate in beneficial or harmful processes. Some SASP-derived factors can stimulate cell proliferation by activation of GRO (growth regulated oncogene) [33,34] and amphiregulin [35]. Other factors triggered by SASP can participate in neovascularization by VEGF activation [36]. Other pathways activated by SASP can produce biphasic WNT activation in cells by SFRP1 (secreted frizzled-related protein 1) [37] as well as IL-6 and IL-8 activation [38,39,40], which in turn can either stimulate or inhibit WNT signaling and cell proliferation, according to the physiological context. Chronic WNT signaling can drive both differentiated cells and stem cells in senescence [41]. Some SASP factors can induce epithelial-to-mesenchymal transition [42]. Other SASP-derived factors as SFRP1 and IL-6 can interfere with stem cell proliferation and modify stem cell niches [43,44,45,46]. Another important effect of SASP-derived factors is initiation and/or potentiation of an inflammatory process. This is of particular relevance for aggravating age-associated diseases [30,31,32,42].
![The numerous activities of the SASP. The many factors that compose the SASP have several biological activities, all highly dependent upon the physiological context. These activities include stimulation of angiogenesis, stimulation or inhibition of cell proliferation, creation of a chemoresistant niche during cancer chemotherapy, stimulation of an epithelial-to-mesenchymal transition, chronic inflammation, alterations of stem cell renewal and/or differentiation, and optimization of tissue repair. Hexagons represent SASP factors that act within and outside the senescent cell. Reproduced with permission from Campisi [11].](https://karger.silverchair-cdn.com/karger/content_public/books/210/parts/chapter_358899/2/m_154037.jpeg?Expires=1716297056&Signature=cZlgxi4MsL0xUKZdTW~ksS9cstr0lCGU8hOg7pth0O5EINw637wIY2rhb-G5VSQWzW1voXTNkGR8SebcYH6OmqG4pwhI0ZnYP6q0pYNOwodohqCTF9EJdemyXYf5u2MSP9gQQjCBjc0Pwm-qCvCWKXNO9lvtWvpL1905p2DHfyOA6-afiiWpHFQTQDByCTIV0lYyOSlt414gTt2ggQ17AcVHQYSeBAt~N31EMe8hMBCv5qrabwPIOnOs4o1hWXOl-3OTQWumDToFz-lC4d7VQdvK1ND1TflMMZeDpCJQyZyqMj6Urn9NW77xlMaaOnMjIQqeYR4sZuMnpRT14DgPkg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
The numerous activities of the SASP. The many factors that compose the SASP have several biological activities, all highly dependent upon the physiological context. These activities include stimulation of angiogenesis, stimulation or inhibition of cell proliferation, creation of a chemoresistant niche during cancer chemotherapy, stimulation of an epithelial-to-mesenchymal transition, chronic inflammation, alterations of stem cell renewal and/or differentiation, and optimization of tissue repair. Hexagons represent SASP factors that act within and outside the senescent cell. Reproduced with permission from Campisi [11].
The numerous activities of the SASP. The many factors that compose the SASP have several biological activities, all highly dependent upon the physiological context. These activities include stimulation of angiogenesis, stimulation or inhibition of cell proliferation, creation of a chemoresistant niche during cancer chemotherapy, stimulation of an epithelial-to-mesenchymal transition, chronic inflammation, alterations of stem cell renewal and/or differentiation, and optimization of tissue repair. Hexagons represent SASP factors that act within and outside the senescent cell. Reproduced with permission from Campisi [11].
As can be deduced from the above summary of SASP-produced effects, it is evident that it represents a highly plastic phenotype. For other details, the extensive review by one of us (J.C.) can be consulted [11].
Senescence-Associated Secretory Phenotype-Triggering Mechanisms and Their Regulation
This property of senescent cells is apparently the result of genomic or epigenetic damage. Cells which enter senescence by other mechanisms, as for instance by p21 or p16INK4a activation, do not exhibit the SASP-phenotype, they just suffer growth arrest [11]. The SASP phenotype is the result of DNA-damage, telomere dysfunction, epigenomic or mitogenic signals, oxidative stress and some other senescence-inducing stimuli [for details see [11]]. This important number of signals inducing the SASP phenotype may explain the variety of its presentation as discussed above. One can assume at this stage of progress on this complicated issue that the activation of the SASP phenotype might ‘serve' to prepare surrounding tissues for repair and/or to engage surrounding cells in clearance of the damaged cells. These functions necessitate underlying regulatory processes. A number (but not all) of SASP processes are positively regulated by the DDR proteins ATM, N8S1 (Nijmegen breakage syndrome 1) CHK2 (checkpoint kinase 2) [22,47,48]. These effects are upstream of p53 which does not regulate positively the SASP phenotype. The stimulating effect of these proteins is conditioned by a previous persistent DDR signaling, in contrast to a rapid transient signaling which does not result in SASP triggering. Persistent DDR signaling develops slowly over several days in culture, after the decrease in the initial DDR signal [22,38]. For the SASP phenotype to develop, DNA-SCARS and TIF are of particular importance. The reason is that these nuclear proteins contain activated DDR proteins necessary for persistent DDR signaling to occur [21]. They are necessary also for senescent growth arrest as well as for the SASP [21,22,38]. Several details of this activation process have still to be clarified. Another important mechanism for the SASP phenotype to develop is the direct activation by the action of nuclear factor-κB (NF-κB) [39,49,50]. C/EBP-β can achieve the same activation [40]. The above transactivators act downstream of signaling cascades controlling proinflammatory cytokine gene expression, especially in immune cells. Early response to senescence-inducing stimuli is activation of expression of the gene coding for IL-1α [51,52]. This cytokine binds its plasma membrane located receptor IL-1R which in turn initiates a signaling cascade activating NF-κB [51,52] which in turn will induce the transcription of genes coding IL-6 and IL-8, both active in mediating inflammation [33,38,39,40]. Growth arrest induced by oncogenes is reinforced and maintained by these cytokines. Contrasting with the positive regulation by DDR, p53 negatively regulates it by inhibiting the SASP phenotype to develop [22,38]. In senescent cells engaged in the SASP phenotype, inactivation of p53 by RNA interference produced an important accentuation of SASP induction essentially by increasing the expression of coding genes [38].
Inactivation of p53 in the absence of p16INK4a expression inducing irreversible growth arrest [39] will result in reinitiation of cell proliferation without however inhibiting SASP expression [22,38]. The occurrence of these cell types is dangerous because by their maintained SASP phenotype they might induce malignancy in neighboring cells.
The Senescent Phenotype and Age-Related Pathologies
The above summarized data are in favor of an important role of the senescent cell phenotype in organismal aging and age-related pathologies. Recent results suggest that the above-defined senescent phenotype does play a role in the development of malignancies. Initially, it was proposed that certain SASP phenotypes may result in inducing the malignant phenotype [30]. In three-dimensional cultures, senescent fibroblasts were shown to disrupt normal tissue structure, especially mammary alveolar and branching morphogenesis and also milk production [53]. This effect of senescent fibroblasts was attributed to the production of MMPs known to be SASP products [33,38]. These effects of senescent fibroblasts can be considered as resulting in the aging of breast structures. Another comparable phenomenon is the stimulation of migration and proliferation of smooth muscle cells (SMCs) of the pulmonary artery by senescent SMCs of the same artery, attributed to IL-6 and IL-8 secretion as well as production of extracellular matrix macromolecules [54]. These processes can lead to the characteristic symptoms of atherosclerosis, intima thickening, medial hypertrophy, resulting in pulmonary hypertension. Comparable pathogenic functions could be attributed to senescent cells in skin aging [12,20,55,56]. Senescent cells and especially some of their products (SASP derived) might contribute to age-dependent thinning of the skin accompanied by loss of collagen, possibly by MMP secretion [for details see [11]]. Another example of the involvement of senescent cells exhibiting the SASP phenotype in age-related diseases concerns the promotion of age-related degenerations, comprising cognitive impairment such as seen in Alzheimer's or Parkinson's disease [57,58]. Similarly, senescent chondrocytes are considered causally involved in osteoarthritits and intervertebral disc degeneration [59,60]. As mentioned above, senescent SMCs but also endothelial cells are involved in the progression of cardiovascular diseases. The role of senescent cells was confirmed in several age-related diseases, as for instance macular degeneration, COPD, emphysema, insulin resistance [11]. Nevertheless, causal relations and underlying mechanisms still have to be elucidated. A transgenic mouse model did however confirm the above relations between senescent cells and age-related pathologies. In these mice, termed INK-ATTAC [61], a promoter element drives the expression of caspase 8 fused to the FK-506 binding protein. The fusion protein dimerizes after the administration of a drug, designated AP20187, thereby activating caspase 8 which induces apoptosis. In this model, cells expressing p16INK4a are eliminated. Such cells are considered as senescent, but more rigorous proof still has to come. When INK-ATTAC mice were crossed with a progeroid mouse expressing constitutively a hypomorphic form of the BubR1 checkpoint protein (BubR1H/H), it resulted in premature aging and death mostly by heart failure. The drug-treated BubR1H/H-INK-ATTAC mice were protected from most age-related pathologies exhibited by the original mouse strain; however, they did not show a significant increase in their life span. They did not suffer from cataract, sarcopenia or loss of subcutaneous fat [61]. Further comparable experiments are still needed before the final acceptance of this attractive hypothesis attributing to senescent cells, especially when exhibiting the SASP phenotype, a role in the pathogenesis of age-related diseases.
Senescent Cells and Cancer
During the early years of the study of senescent cell phenotype, the dominant paradigm was that mechanisms at work at entering senescence provided protection against malignant transformation [6,7,8]. This theory and arguments favoring it were cited in an extensive review on the effect of cell-matrix interactions and the malignant process [8].
As proposed than by J.C.: ‘most senescence inducing stimuli converge on two cancer protective gene expression pathways related either to p53 or pRB' [6,7]. Among the mechanisms mentioned was the loss of p53 which abrogates cellular senescence at least in cells expressing p21 but not in those expressing p16. Senescent cells express usually one or the other of these genes. Some strains of cells reaching replicative senescence are either p16 or p21 positive. The p16-expressing cells do not present loci with markers of DNA damage. This might be one possible mechanism linking the senescent phenotype with increased oncogenic risk, through p53 dysfunction. Several reports confirmed the suppression of cancer development by entering senescence [6,8,62]. In human cells, IL-6, IL-8 and IGFBP7 (insulin-like growth factor binding protein 7) stabilize the senescent phenotype induced by the oncogenic forms of RAS or BRAF [39,40]. RAS and BRAF are cytoplasmic proteins that participate in signal transductions by growth factors and extracellular signals to the cell interior. The genes coding for these proteins are frequently mutated in human cancers. GROα, a potent mitogen, also an SASP component, is induced by oncogenic RAS; it promotes the senescence of normal human ovarian fibroblasts [34]. It appears that several SASP factors, among them IL-6, IL-8, IGBP7 and GROα contribute to the establishment of the oncogene-induced senescent phenotype. IL-6 and IL-8 act by activating an intracellular feedback loop contributing to activate NF-κB and C/EBP-β transcription factors [39,40]. Secreted WNT16B is an important stabilizer of the senescent growth arrest of human fibroblasts [62].
There is recent evidence that senescent cells can also act by favoring the malignant phenotype, according to gene expressions driven by specific environmental conditions. One argument in favor of this more recent contention comes from studies on xenografts. Coinjection of senescent fibroblasts stimulated the proliferation of mouse and human epithelial tumor cells in immunocompromised mice [36,63,64]. Soluble factors derived from senescent cells are involved [63]. Two of these factors are MMP-3 (stromelysine) [64] and VEGF [36] involved also in tumor angiogenesis. Amphiregulin and GROs [33,35] as well as a number of other substances, also derived from SASP, also stimulate epithelial to mesenchymal transition (fig. 3). Other SASP factors as IL-6 and IL-8 secreted by senescent fibroblasts also contribute to the epithelial-mesenchymal transition and the development of the malignant phenotype [38,42,53].
It appears from the forgoing that the age-dependent accumulation of senescent cells might create a microenvironment favoring malignant transformation and progression. Among the above-mentioned factors, it is probably the initiation of inflammation and contribution to DNA damage creation which are the most important. This represents a real inversion of the previous paradigm attributing to senescence a way out to avoid malignant transformation. It seems probable that both possibilities coexist and the final issue depends on the predominance of some of the factors such as SASP products pushing the process in one way or the other. Among these possibilities, the one offering protection to malignant cells from therapeutic agents by the secretion by DNA-damaged senescent cells, WNT1GB, IL-6 and TIMP-1, all SASP derivatives, is the most surprising [65,66]. Some other SASP elements, on the contrary, might be chemosensitizing. Global suppression of SASP by NF-κB inhibition promoted resistance to chemotherapy in a mouse lymphoma model [67]. It is therefore important to combine chemotherapy with adjuvant therapy in order to eliminate senescent cells from both the host tissue and the tumor in order to avoid the above-described protective effects of senescent cells decreasing the efficiency of chemotherapy.
The above results are also in sharp contradiction with the initially proposed ‘beneficial' effect for cells enabling them to escape malignant transformation by ‘entering senescence' [6,7]. As a matter of fact, this originally emphasized capacity of ‘escaping' malignant transformation thanks to the senescent phenotype remains however valid also. Here again, certain forms of the SASP reaction do play a role by stabilizing growth arrest [15,68,69]. In human cells, IL-6, IL-8 and IGFBP7 stabilize growth arrest induced by the oncogenic forms of RAS and BRAF [39,40,70]. These two are cytoplasmic proteins active in transducing signals from growth factors to the cell interior and, as mentioned, are frequently mutated in human cancers. GROα, also an SASP product with potent mitogenic capacity, induced also by oncogenic RAS, promotes senescence of normal human ovarian fibroblasts [34]. It appears therefore that at least some of the SASP components act by creating a self-sustaining signaling feedback loop which stabilizes the senescent phenotype by activating the NF-κB and C/EBP-β transcription factors [39,40]. Secreted WNT16B in human and mouse cells stabilize the senescent growth arrest by the intermediary of a RAS oncogene [48]. These examples confirm previous findings showing that in some specific circumstances the senescent phenotype by its SASP components helps to ‘escape' malignancy by the stabilization of the senescent phenotype.
Immune Clearance
By the proinflammatory activity of several SASP components, senescent cells were shown to interfere with immune cell activity [38,49,50]. One of their functions, which may be reinforced in the clearance of senescent cells, is a sort of induced suicide. But another reinforced effect appears to be the elimination of oncogene-expressing cells; thus, these activities concern both senescent cells as well as oncogene-transformed cells. By these activities, senescent cells can induce a protective effect against pre-malignant and malignant cells. The action of senescent cells concerns several types of immune cells, natural killer (NK) cells, macrophages and T cells [38,39,50]. In case of genomic damage, the SASP reaction can induce the expression of the membrane-bound ligand for the NK receptor NKG2D [51]. These protective effects of the immune system against the senescent cell-induced damage to the organism decline however with age, as discussed by T. Fulop (this vol.). Another possibility for the age-dependent increase in the number of senescent cells with age is the probability of increased damage frequency and severity, oncogenic mutations and other senescence-inducing events. The previously cited experiments show that the time factor alone is enough to produce an increase in senescent cells, at least in culture. Aged tissues exhibit an increased number of cells with DNA damage foci as seen also in senescent cells [23,24,71]. Another contributing fact is the SASP-induced senescence of cells which evade immune clearance [31,32] perhaps by the increased level of MMP secretion which can attack cell surface receptors and ligands on immune cells, protecting thereby senescent cells and preventing their elimination.
Tissue Repair
Recent experiments showed that SASP associated with the senescent phenotype may facilitate the repair of some damaged tissues [30,51,52,53]. In a mouse model, acute liver injury induced the senescence of hepatic stellate cells followed by their clearance mediated essentially by NK cells [39]. In another mouse model where the animals were devoid of both the p53/p21 and p16INK4a/pRB pathways, eliminating the senescent response, wound healing was tempered by an excess of fibrotic response [39]. These findings are in agreement with previous reports showing that senescent hepatic stellate cells increased the inflammatory response but decreased fibrosis [71].
In another experiment on skin wounding in mice, injury induced cell senescence in fibroblasts by a signaling cascade initiated by CCN1, a component of the extracellular matrix by its binding to a cell membrane-bound integrin [54]. This integrin-mediated signaling induces senescent growth arrest as well as increased production of several SASP proteins. In mice expressing a mutated form of CCN1 deficient in integrin binding and so incapable of inducing senescence in wounds lacking senescent cells, their healing was again accompanied by increased fibrosis [54]. These experiments, on two different locations of tissue repair in mice, the liver and the skin, show that SASP mediated by senescent cells can normalize wound healing and prevent fibrosis. How come than that tissue repair declines with age in presence of an increasing number of senescent cells? One possibility is the transitory presence of senescent cells in younger animals, where they might well have been cleared soon after their SASP-mediated favorable effect on wound healing. This is no more the case in aging organisms where the number of senescent cells increases, not being anymore cleared by the above-described mechanism. Clearly, more experiments on this important issue of cell-tissue aging are needed.
Theoretical Considerations
We attempted to show in this chapter the long way travelled since the initiation of cell cultures for the study of aging. These experiments suffer from an evident shortcoming or bias: can the cultured cell, after in vitro aging of some selected cell types, yield significant information on the aging of the organism? Against numerous critics raised against the Hayflick model, his method was adopted by a large number of laboratories all over the world. The essential conclusions of the Hayflick model are the limited proliferation capacity of normal, nontransformed (nonmalignant) cells which after a limited number of divisions (doubling of cells in culture) stop dividing, and adopt characteristics of a ‘senescent cell' (see fig. 3). This cell culture-produced paradigm is currently attributed to a loss of telomeres by repression of the telomerase system capable to keep their critical length compatible with further proliferation. These conclusions by Hayflick, confirmed by a number of laboratories, led to what we called the Hayflick paradigm, but this also revealed several problems occurring during repeated rounds of proliferation, as discussed by Macieira-Coelho in another chapter. For a while, little attention was paid to the senescent state reached by repeated cell divisions. It was known that cells at the end of their proliferative life are not dead. They exhibit clear signs of ‘cell aging', pyknotic nuclei, vacuolated cytoplasm, altered mitochondria, lipofuscin accumulation and others [4]. It was up to a new generation to take up this challenge; it was met by one of us (J.C.) and resulted in a number of interesting observations concerning the senescent phenotype. The first results pointed to the possibility of reaching the senescent phenotype without going all through the ‘permitted' number of cell duplications. This short cut was shown to be mediated by antioncogene-initiated pathways, as described above. This subject was then progressively taken up by a number of laboratories as shown by (the limited number of) references. Methods as staining for SA-β-Gal and others, cited above, enabled the demonstration of senescent cells in tissues, also their increase with age as well as in malignant tumors. This chapter presents a summary of some of the most important recent results. It is highly probable that more teams will produce more experimental results on this new and exciting topic in a near future.
Among the questions that remain to be answered is the relation between the senescent cells as described by Hayflick and followers [1,2,3,4] and the senescent cells produced by the antioncogene-mediated pathway, as described above. Further work will be clearly needed to answer this question.
