

Abstract
Glucocorticoids are essential steroid hormones secreted from the adrenal gland in response to stress. Since their discovery in the 1940s, glucocorticoids have been widely prescribed to treat inflammatory disorders and hematological cancers. In the traditional view, glucocorticoids are regarded as anti-inflammatory molecules; however, emerging evidence suggests that glucocorticoid actions are more complex than previously anticipated. The anti-inflammatory activity of glucocorticoids is attributed to the repression of pro-inflammatory genes through signal transduction by their steroid receptor, the glucocorticoid receptor (GR). The mechanisms modulating the pro-inflammatory effects of glucocorticoids are not well understood. In this review, we discuss recent findings that provide insights into the mechanism by which GR signaling can play a dual role in the regulation of the immune response. We hypothesize that these apparently opposite processes are working together to prepare the immune system to respond to a stressor (pro-inflammatory effects) and subsequently restore homeostasis (anti-inflammatory effects). Finally, we propose that determining the mechanisms which underlie the tissue-specific effects of glucocorticoids will provide an excellent tool to develop more efficient and selective glucocorticoid therapies.
Glucocorticoids and the Stress Response
Glucocorticoids are steroid hormones synthesized and secreted by the adrenal gland in response to stress [1]. Upon exposure to stress, the hypothalamus is stimulated to release corticotrophin-releasing hormone, which then acts on the anterior pituitary gland to stimulate the synthesis of adrenocorticotropic hormone (ACTH). ACTH then acts on the adrenal cortex to induce the secretion of glucocorticoids [2]. Once in circulation, glucocorticoids exert a variety of tissue-specific effects (fig. 1) [2,3,4,5]. Therefore, glucocorticoid imbalances can result in pathological conditions such as the severe cardiovascular, metabolic and immunological complications observed in Cushing's syndrome (glucocorticoid excess) and Addison's disease (glucocorticoid deficiency).
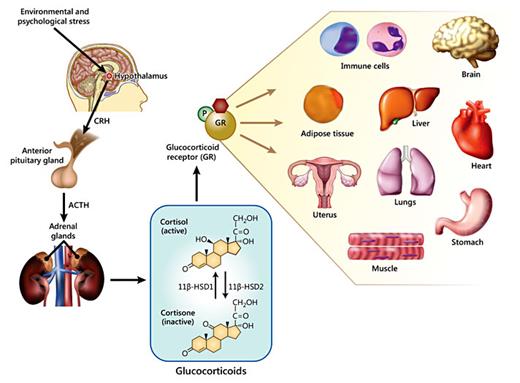
Regulation of glucocorticoid secretion in response to stress by the hypothalamic-pituitary-adrenal axis. Upon exposure to environmental or psychological stress the hypothalamus is stimulated to release corticotropin-releasing hormone (CRH). CRH then stimulates the anterior pituitary gland to secrete ACTH. In turn, ACTH targets the cortex of the adrenal glands to release cortisol into the bloodstream. Once in circulation, cortisol can be converted to the inactive form, cortisone, by 11β-hydroxysteroid dehydrogenase type 2. Conversely, 11β-hydroxysteroid dehydrogenase type 1 converts cortisone to cortisol. Glucocorticoids exert their effects by binding to their receptor, the GR. GR is expressed in virtually all cell types and tissues. Thus, GR signaling plays an important role in the modulation of a large number of biological functions in immune cells and in several organs and tissues, including the brain, liver, heart, lungs, adipose tissue, reproductive system, stomach and muscle.
Regulation of glucocorticoid secretion in response to stress by the hypothalamic-pituitary-adrenal axis. Upon exposure to environmental or psychological stress the hypothalamus is stimulated to release corticotropin-releasing hormone (CRH). CRH then stimulates the anterior pituitary gland to secrete ACTH. In turn, ACTH targets the cortex of the adrenal glands to release cortisol into the bloodstream. Once in circulation, cortisol can be converted to the inactive form, cortisone, by 11β-hydroxysteroid dehydrogenase type 2. Conversely, 11β-hydroxysteroid dehydrogenase type 1 converts cortisone to cortisol. Glucocorticoids exert their effects by binding to their receptor, the GR. GR is expressed in virtually all cell types and tissues. Thus, GR signaling plays an important role in the modulation of a large number of biological functions in immune cells and in several organs and tissues, including the brain, liver, heart, lungs, adipose tissue, reproductive system, stomach and muscle.
Glucocorticoid therapy was first introduced by Dr. Philip Hench in the 1940s for the treatment of rheumatoid arthritis [6]. Since then, glucocorticoids have commonly been prescribed to treat inflammatory disorders, including asthma, allergic rhinitis, ulcerative colitis, and several other dermatological, ophthalmic, neurological and autoimmune diseases [7,8]. Despite their therapeutic benefits, glucocorticoid use, in traditional high doses >5 mg/day, is associated with severe side effects, including diabetes, hypertension, glaucoma, muscle atrophy and growth retardation [7,9]. However, the magnitude of the positive or negative effects of glucocorticoids will depend on the dose, duration of the treatment, glucocorticoid receptor (GR) levels, and cell- and tissue-specific glucocorticoid signal transduction [10,11,12].
Long-term treatment with glucocorticoids can also be associated with glucocorticoid resistance, which results in the inability of glucocorticoids to exert their effects on target tissues, limiting the efficacy of the therapy [13]. Therefore, understanding how glucocorticoids exert their actions in a dose- and tissue-specific manner should help to develop novel therapies using these agents that may decrease their undesired effects. This review examines evidence that emphasizes the pro- and anti-inflammatory actions of glucocorticoids on the immune system. These apparently opposite effects appear to work together as a defense mechanism to ‘prepare' the immune system to respond to a stressor, and subsequently to shut down the immune response to restore homeostasis [14].
Molecular Mechanisms of Glucocorticoids
GR Structure
Glucocorticoids exert their actions by binding to their receptor, the GR, a ligand-induced transcription factor and member of the nuclear receptor superfamily [15]. GR is expressed in virtually all cell types and tissues [1,3,8]. In terms of the structure, GR is composed of 3 major functional domains: the N-terminal domain (NTD); the central DNA-binding domain (DBD); the C-terminal ligand-binding domain (LBD), as well as a hinge region (region linking the DBD and LBD; fig. 2a) [2,14]. Each domain comprises a specific function; for example, the NTD has a transcriptional activation function (AF)-1 domain which contains the majority of the residues subject to post-translational modifications (PTMs). The NTD is important for the recruitment of the basal transcriptional machinery. The DBD consists of two zinc fingers implicated in DNA binding, nuclear translocation and GR dimerization [2]. The LBD also contains an AF-2 domain which interacts with co-regulators in a ligand-dependent manner [14,16,17,18].
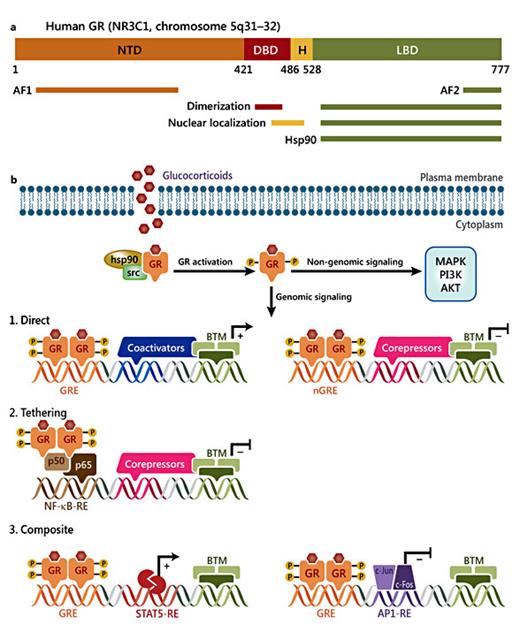
Schematic representation of the GR structure and signaling pathways. a GR domains and regions involved in transactivation (AF1 and AF2), dimerization, nuclear localization and Hsp90 binding. H = Hinge region. b GR can signal in a non-genomic manner by modulating the activity of several kinases, including MAPK, phosphoinositide 3-kinase (PI3K) and AKT. GR regulates gene expression by three mechanisms: (1) direct - activated GR binds to GREs or nGREs on the promoter or sequence of target genes; (2) tethering - GR tethers itself to other DNA-bound transcription factors; (3) composite - GR binds directly to a GRE and interacts with neighboring DNA-bound transcription factors. BTM = Basal transcription machinery; STAT = signal transducer and activator of transcription.
Schematic representation of the GR structure and signaling pathways. a GR domains and regions involved in transactivation (AF1 and AF2), dimerization, nuclear localization and Hsp90 binding. H = Hinge region. b GR can signal in a non-genomic manner by modulating the activity of several kinases, including MAPK, phosphoinositide 3-kinase (PI3K) and AKT. GR regulates gene expression by three mechanisms: (1) direct - activated GR binds to GREs or nGREs on the promoter or sequence of target genes; (2) tethering - GR tethers itself to other DNA-bound transcription factors; (3) composite - GR binds directly to a GRE and interacts with neighboring DNA-bound transcription factors. BTM = Basal transcription machinery; STAT = signal transducer and activator of transcription.
GR Genomic and Non-Genomic Effects
Under basal conditions, GR resides primarily in the cytoplasm in a complex with chaperone proteins hsp90, hsp70 and p23, and immunophilins FKBP51 and FKBP52, where it is largely considered to be functionless [2,5]. Upon hormone binding, GR translocates into the nucleus to enhance or repress the transcription of target genes by several mechanisms: (1) direct binding to specific cis-acting DNA sequences (glucocorticoid-responsive elements; GREs); (2) tethering itself to other transcription factors, and/or (3) through direct binding to DNA and interacting with neighboring transcription factors, which is known as composite regulation (fig. 2b) [2].
The GRE consensus sequence is GAGAACAnnnTGTTCT, an imperfect palindrome containing two hexameric half sites separated by three base pairs [18,19,20]. GR binding to GREs typically leads to transactivation, where glucocorticoids induce target genes. However, recent studies have shown that GR occupancy of a GRE can also lead to gene repression, in a process known as ‘transrepression' (fig. 2b) [21]. GR can also repress genes by tethering itself to other transcription factors. For example, GR can physically interact with nuclear factor-κB (NF-κB) and activator protein-1 (AP-1), which represses their capacity to induce the transcription of pro-inflammatory genes (fig. 2b) [22,23]. GR is also known to bind to inverted palindromic sequences denominated as negative GREs (nGREs). The consensus sequence of these cis-acting negative response elements is CTCC(n)0-2GGAGA [24]. Studies have shown that GR recruitment to nGREs promotes the assembly of a corepressor complex and the recruitment of histone deacetylases, which direct glucocorticoid-dependent repression of specific genes [25] (fig. 2b).
GR has also been proposed to exert ‘rapid' non-genomic actions by directly modulating signal transduction pathways [2]. This process occurs via membrane-bound GR or cytosolic GR interactions with kinases, such as the ERKs (extracellular signal-regulated kinases), the JNKs (c-Jun NH2-terminal kinases), the p38s (p38 isoforms), and ERK5 [26] (fig. 2b). To date, the detailed mechanism and biological implications of non-genomic effects of GR have not been fully elucidated; human studies suggest that this response might have important physiological effects on the cardiovascular and immune system [26,27]. Non-genomic effects of glucocorticoids are extensively discussed in a recent publication by Ayroldi et al. [26].
GR Isoforms
In addition to the genomic and non-genomic actions of glucocorticoids, GR signaling is also dependent on the existence of multiple receptor isoforms and PTMs [2,4]. GR is transcribed from a single gene, NR3C1. However, alternative splicing of this gene generates GRα and GRβ isoforms, which differ in their C-terminal domain [28]. Both isoforms are associated with distinct responses to glucocorticoids [29]. The existence of different isoforms explains in part how a ‘single' receptor can exert a plethora of pharmacological and cellular responses. Human GRα, the classical GR protein, exerts most of the biological actions of glucocorticoids [29]. Furthermore, GRα expression is higher than GRβ in tissues [30]. In general, GRβ presents several distinct properties: it is located primarily in the nucleus, does not bind glucocorticoid agonists, and antagonizes the activity of GRα [31,32,33]. Studies have shown that increased GRβ levels are associated with glucocorticoid resistance in inflammatory disorders, including asthma, rheumatoid arthritis and ulcerative colitis [34]. However, genome-wide microarray studies on cells selectively expressing GRβ have shown that GRβ regulates the expression of a large number of genes independently of GRα activity [34,35]. These findings raise interesting possibilities regarding GRβ genomic effects and the physiological role of GRβ, although the molecular mechanisms governing GRβ actions are still unknown. In addition to GRβ, three less-well-characterized isoforms have been reported: GRγ, GR-A and GR-P [31]. Although their physiological role is poorly understood, studies suggest that their expression could be associated with the development of glucocorticoid resistance in some types of hematological cancers [31].
GRα and GRβ can also undergo alternative translation initiation in exon 2, which generates several GR isoforms with distinct properties. Eight additional isoforms of GR with truncated N-terminal are generated from GRα: GRα-A, GRα-B, GRα-C1, GRα-C2, GRα-C3, GRα-D1, GRα-D2 and GRα-D3. Similar isoforms can also be generated from GRβ [2]. The structure, complexity and signaling properties of the GR isoforms are discussed in detail in a recent review by Oakley and Cidlowski [2].
GR PTMs
PTMs lead to important changes in the transcriptional activity of the GR. The most frequently studied PTM of GR is phosphorylation [31]. Human GR has been reported to be phosphorylated at several serine residues (S113, S134, S141, S143, S203, S211, S226 and S404) by various kinases, including mitogen-activated protein kinase (MAPK), GSK-3 and cyclin-dependent kinase [36,37]. Phosphorylation of GR has been shown to impact its activity at multiple levels.
Studies in COS-1 cells transfected with phosphorylation-deficient GRα mutants showed that changes to the phosphorylation status of GR blocks the ability of the receptor to activate GR target genes [38]. Phosphorylation of GR at S211, a substrate for p38 MAPK, is associated with increased GR-induced gene transcription [39]. In contrast, phosphorylation of S226 is associated with decreased GR signaling transduction [40]. Phosphorylation of GR S404 has also been shown to drive a specific transcriptional response to glucocorticoids [41]. The phosphorylation state of GR influences its response to hormone, cellular localization and signaling activity. Furthermore, recent studies have shown that S134-GR residue can be phosphorylated by p38 MAPK in response to stress in the absence of hormone binding [42]. This study showed that mutations of Ser134 (S134A-GR) lead to the dysregulation of genes associated with metabolic and inflammatory diseases [42], which indicate that hormone-independent phosphorylation events are also critical in the regulation of GR-dependent gene transcription.
Other PTMs have also been reported to modulate GR activity and signaling. Ubiquitination of GR at Lys-419 targets the receptor for proteasomal degradation [43]. SUMOylation and acetylation also play a role in the modulation of GR transcriptional activity by enhancing or repressing its interactions with specific co-regulators [44]. For example, deacetylation of GR Lys-494 and Lys-495 residues has been reported to modulate GR repression of NF-κB [45]. In addition, studies suggest that acetylation of GR modulates the ability of the receptor to induce or repress glucocorticoid-responsive genes in target tissues [46]. In summary, the existence of multiple GR isoforms and the presence of numerous PTMs contribute to the diversity of the genomic and non-genomic actions of GR, and perhaps explain how a single receptor can exert a plethora of physiological actions.
Anti- and Pro-Inflammatory Effects of Glucocorticoids
The discovery of the anti-inflammatory actions of glucocorticoids was a major breakthrough for the treatment of inflammatory disorders. Both natural and synthetic glucocorticoids are widely prescribed as anti-inflammatory drugs [3,14]. The anti-inflammatory activity of GR and glucocorticoids generally is attributed to the repression of pro-inflammatory genes through the direct interaction of GR with other transcription factors [21]. However, anti-inflammatory activity of GR can also result via non-specific interactions of glucocorticoids with membrane components or through membrane-bound GR [2]. In addition, GR interactions with kinases can also affect inflammatory signaling pathways independently of gene transcription [14,26].
Although glucocorticoid actions are typically described as anti-inflammatory, studies suggest that glucocorticoids can also exert pro-inflammatory effects in response to acute stress [47]. For example, glucocorticoid treatment can exacerbate the peripheral immune response in delayed-type hypersensitivity [47,48,49]. Also, the levels of pro-inflammatory cytokines such as interleukin (IL)-1β have been found to be increased in the central nervous system in response to acute stress associated with increased glucocorticoid secretion [47,50,51]. In addition, recent studies suggest that chronic exposure to glucocorticoids, classically viewed as anti-inflammatory, may result in increased systemic trafficking of lymphocytes and monocytes [52]. In the next sections we will review how glucocorticoids modulate inflammation by suppressing or augmenting the immune response.
Classic Anti-Inflammatory Effects of Glucocorticoids
As noted above, the actions of glucocorticoids are classified as anti-inflammatory/immunosuppressive. Glucocorticoids suppress inflammation by multiple mechanisms that impact both the innate and adaptive immune responses [14].
The innate immune system is the first line of defense in response to a pathogen or injury. This response is activated by the recognition of PAMPs (pathogen associated molecular patterns) or DAMPs (damage-associated molecular patterns) through invariant pattern recognition receptors. These receptors are expressed on the surface of macrophages, dendritic cells, histiocytes, Kupffer cells and mastocytes [14,53]. The initiation of the immune response is characterized by the activation of the complement cascade, recruitment of immune cells, production of cytokines and chemokines, and ultimately activation of the adaptive immune system [53].
The adaptive immune response is a second line of defense composed of highly specialized processes mediated by T and B lymphocytes. These cells are derived through hematopoiesis from committed progenitor cells before differentiating into their distinct lymphocyte types. Newly formed lymphocytes then migrate to peripheral lymphoid tissues and enter into circulation, where they survey and eliminate specific pathogens. Following antigen neutralization, the surviving cells become memory cells that remain in circulation with capacity to respond to the same antigen upon re-exposure [54].
Glucocorticoids regulate the immune response at both the cellular and transcriptional level. At the cellular level, glucocorticoids can induce apoptosis of T lymphocytes, neutrophils, basophils and eosinophils to reduce inflammation [47]. In a lipopolysaccharide (LPS)-induced sepsis model, glucocorticoids were shown to modulate macrophage cytokine production by inhibiting p38 MAPK [14,55]. Glucocorticoids also suppress inflammation in allergy disorders by regulating the expression of IL-1β, monocyte chemoattractant protein-1, macrophage inflammatory protein-2 and interferon-γ-inducible protein 10 in neutrophils and macrophages [14,56]. Thus, the primary anti-inflammatory action of glucocorticoids is to repress a plethora of pro-inflammatory genes encoding cytokines, chemokines, cell adhesion molecules, inflammatory enzymes and receptors to resolve the inflammatory process and restore homeostasis (fig. 3). The mechanisms involved in GR repression of the transcriptional activity of AP-1 and NF-κB have been a primary topic of research as a molecular mechanism driving the anti-inflammatory properties of GR [7,14,57].
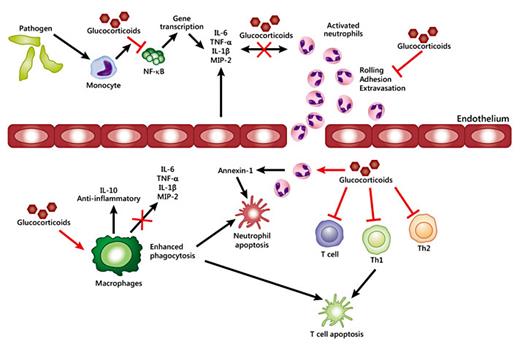
Anti-inflammatory effects of glucocorticoids. Exposure to pathogens leads to a fast activation of the immune response. Glucocorticoids modulate the inflammatory response by repressing the expression of pro-inflammatory cytokines by immune cells. In addition, glucocorticoids can repress the expression of adhesion molecules, which prevents rolling, adhesion and extravasation of neutrophils to the site of inflammation. Glucocorticoids also induce the expression of annexin-1. Synthesis of annexin-1 promotes neutrophil detachment and apoptosis. Chronic exposure to glucocorticoids induces a switch in resident macrophages gene expression profile from pro- to anti-inflammatory, and increases macrophages phagocytic activity. Finally, glucocorticoids act on T cells by blocking Th1- and Th2-derived cytokine production and inducing cell death.
Anti-inflammatory effects of glucocorticoids. Exposure to pathogens leads to a fast activation of the immune response. Glucocorticoids modulate the inflammatory response by repressing the expression of pro-inflammatory cytokines by immune cells. In addition, glucocorticoids can repress the expression of adhesion molecules, which prevents rolling, adhesion and extravasation of neutrophils to the site of inflammation. Glucocorticoids also induce the expression of annexin-1. Synthesis of annexin-1 promotes neutrophil detachment and apoptosis. Chronic exposure to glucocorticoids induces a switch in resident macrophages gene expression profile from pro- to anti-inflammatory, and increases macrophages phagocytic activity. Finally, glucocorticoids act on T cells by blocking Th1- and Th2-derived cytokine production and inducing cell death.
AP-1 is an important regulator of gene expression and contributes to the control of many cytokine genes. A recent review by Busillo and Cidlowski [14] elegantly described the mechanism of GR-mediated inhibition of AP-1. AP-1 is generally activated in response to environmental stress, such as radiation, and growth factors [58]. AP-1 can function as a homo- or heterodimer, and the heterodimer subunit composition is variable. AP-1 heterodimer can be formed with the basic leucine-zipper transcription factors Fos (cFos, Fos B, Fra-1 and Fra-2), Jun (c-Jun, v-Jun, Jun B and Jun D), activating transcription factor (ATF2, ATF3, B-ATF, JDP-1 and JDP-2) or MAF (V-maf musculoaponeurotic fibrosarcoma oncogene homolog: MAFA, MAFB, c-MAF, NRL, MAFF, MAFG and MAFK) [14]. The most common form of the AP-1 heterodimer is the c-Fos/c-Jun heterodimer [59]. GR-mediated inhibition of AP-1 is achieved by three mechanisms (fig. 4): (1) binding to a GRE on the promoter and tethering to c-Jun to repress the transcriptional activity of AP-1; (2) direct interaction of GR (tethering) with the c-Jun subunit of AP-1, and (3) induction of MKP-1 to inactivate JNK, which in turn decreases c-Jun transcriptional activity.

Glucocorticoids exert anti-inflammatory effects by repressing the expression of AP-1 and NF-κB. Exposure to pathogens triggers a signaling cascade that leads to the activation of pro-inflammatory transcription factors, including AP-1 and NF-κB. Activation of GR results in its translocation to the nucleus. Once in the nucleus, GR represses AP-1 and NF-κB. GR can repress AP-1 by the following mechanisms: (i) in some promoters, GR binds to a GRE and simultaneously interacts with c-Jun to repress AP-1 activity; (ii) GR can also physically interact (tethering) with c-Jun, which represses AP-1 activity and the transcription of inflammatory genes. NF-κB activity can be repressed by GR through the following mechanisms: (i) GR can physically interact with p65, which represses the activity of NF-κB; (ii) GR can recruit GRIP (GR interacting protein) which blocks the formation of the NF-κB /IRF3 heterodimer; (iii) GR can prevent the phosphorylation and activation of RNA polymerase II (Pol II) by blocking the recruitment of pTEFb (positive transcription elongation factor); (iv) GR is also able to repress NF-κB by recruiting HDAC (histone deacetylases); (v) GR prevents NF-κB from interaction with p300 and CPB (CREB1-binding protein); (vi) GR can interact with p53, which alters NF-κB pro-inflammatory transcriptional activity.
Glucocorticoids exert anti-inflammatory effects by repressing the expression of AP-1 and NF-κB. Exposure to pathogens triggers a signaling cascade that leads to the activation of pro-inflammatory transcription factors, including AP-1 and NF-κB. Activation of GR results in its translocation to the nucleus. Once in the nucleus, GR represses AP-1 and NF-κB. GR can repress AP-1 by the following mechanisms: (i) in some promoters, GR binds to a GRE and simultaneously interacts with c-Jun to repress AP-1 activity; (ii) GR can also physically interact (tethering) with c-Jun, which represses AP-1 activity and the transcription of inflammatory genes. NF-κB activity can be repressed by GR through the following mechanisms: (i) GR can physically interact with p65, which represses the activity of NF-κB; (ii) GR can recruit GRIP (GR interacting protein) which blocks the formation of the NF-κB /IRF3 heterodimer; (iii) GR can prevent the phosphorylation and activation of RNA polymerase II (Pol II) by blocking the recruitment of pTEFb (positive transcription elongation factor); (iv) GR is also able to repress NF-κB by recruiting HDAC (histone deacetylases); (v) GR prevents NF-κB from interaction with p300 and CPB (CREB1-binding protein); (vi) GR can interact with p53, which alters NF-κB pro-inflammatory transcriptional activity.
NF-κB is also involved in the transcriptional regulation of genes involved in inflammation. NF-κB is composed of five members: NF-κB1 (p50/p105), NF-κB2 (p50/p100), RelA (p65), c-Rel, and RelB [60]. Inactive NF-κB complexes are held in the cytoplasm via their non-covalent interaction with an inhibitory protein known as IκB [60]. Upon activation, the NF-κB/IκBα complex is phosphorylated and IκB is degraded, which leads to NF-κB release and translocation to the nucleus. Once in the nucleus, NF-κB binds to specific DNA-binding sites (5′-GGGRNNYYCC-3′) in the promoter or enhancer regions of numerous pro-inflammatory genes [60]. The mechanisms by which GR represses NF-κB activity are similar to those proposed for AP-1 (fig. 4). GR can physically interact with RelA [61], and can also sequester the GR interaction protein, GRIP-1, away from interferon regulatory transcription factor 3 (IRF3), which blocks the formation of the p65/IRF3 complex. GR is then tethered to the NF-κB complex leading to the transrepression of cytokines and chemokines [62].
GR can also repress NF-κB activity by recruiting histone deacetylases to NF-κB-dependent promoters [45]. GR binding to CREB-binding protein and p300 [63], as well as GR interfering with serine-2 phosphorylation at the C-terminal domain of RNA polymerase II [64], are general mechanisms by which GR is able to regulate NF-κB activity negatively. Recent studies also suggest that GR interplay with the p53 signaling is important to control inflammation via GR transrepression of NF-κB [65].
GR can also regulate the inflammatory response by inducing the expression of proteins that can block pro-inflammatory pathways. Glucocorticoids have inhibitory effects on the MAPK signaling pathways through MAPK phosphatase-1, which inhibits p38 MAPK, preventing the induction of multiple inflammatory genes [14]. In addition to this pathway, molecular evidence suggests that glucocorticoids influence the mRNA stability of inflammatory genes by regulating the expression of tristetraprolin (TTP) [66]. TTP plays an important role in the resolution phase of the inflammatory response by destabilizing the mRNA of many pro-inflammatory cytokines and targeting them for degradation [67], and dexamethasone (a synthetic glucocorticoid) treatment of A549 lung epithelial cells induces TTP gene expression [66]. These studies provide evidence that glucocorticoids can repress the inflammatory process indirectly via the induction of anti-inflammatory molecules. In summary, glucocorticoids can modulate inflammation directly at the transcriptional level by repressing the gene expression of pro-inflammatory molecules, or through post-transcriptional mechanisms via interactions with anti-inflammatory proteins.
Pro-Inflammatory Effects of Glucocorticoids
Glucocorticoid secretion in response to stress has been assumed mainly to be anti-inflammatory, as discussed above. However, studies suggest that the type of exposure to glucocorticoids and the basal state of the immune system are important factors influencing the effects of glucocorticoids [47,48,50,51,52,68]. For example, while chronic exposure to glucocorticoids seems to be immunosuppressive, acute exposure enhances the peripheral immune response [48]. The mechanisms by which the same hormone directs opposite responses are not well understood.
Genome-wide microarray studies of human mononuclear cells suggest that glucocorticoids can induce the expression of several innate immune-related genes, including several members of the Toll-like receptor (TLR) family [69,70]. However, in the same cells, glucocorticoids have an inhibitory effect on the expression of pro-inflammatory genes of the adaptive immune response [70]. These findings are interesting because it is generally accepted that glucocorticoids suppress TLRs - the activation of which is a hallmark feature of inflammation - through the induction of MKP-1 or glucocorticoid-induced leucine zipper, or via repression of AP-1 and NF-κB [69].
GR signaling interplays with TLR signaling pathways through several mechanisms [14]. For the purpose of this review, we will discuss briefly the mechanisms that enhance the activity of TLRs (fig. 5a). Several studies have reported that glucocorticoids induce the expression of TLR2 and TLR4 in lung epithelial cells [69,71,72]. Interestingly, glucocorticoids have also been shown to increase Haemophilus influenzae-induced expression of TLR2 mRNA and protein levels via negative cross-talk with p38 MAPK [73]. A study by Hermoso et al. [72] showed that dexamethasone enhances TNF-α induction of TLR2 via the activation of GR.
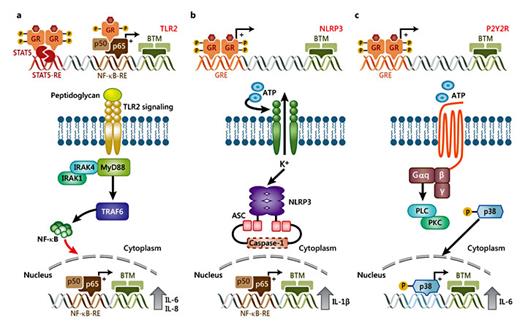
Glucocorticoid-mediated modulation of pro-inflammatory pathways. a GR can drive the expression of TLR2 by interacting with STAT5 and NF-κB. TLR2 recognizes the bacterial cell wall component peptidoglycan. Upon stimulation, MYD88 binds to the cytoplasmic portion of TLR2. This event leads to recruitment and activation of IRAK1, IRAK4 and TRAF6, ultimately leading to the downstream signaling activation of pro-inflammatory transcription factors which drives the expression of inflammatory cytokines, including IL-6 and IL-8. b, c Glucocorticoids regulate the expression of NLRP3 and P2Y2R by mechanisms that are not well understood. NLRP3 regulates the immune system response to injury or pathogens by sensitizing macrophages to extracellular ATP (danger signal) and inducing the synthesis of IL-1β. P2Y2R is a G-protein-coupled receptor that is activated in response to ATP, which stimulates the activation of PLC (phospholipase C) and PKC (protein kinase C), and the subsequent downstream signaling. Glucocorticoids can upregulate the expression of P2Y2R by enhancing the ATP-dependent activation of p38, which leads to the expression of the pro-inflammatory cytokine IL-6.
Glucocorticoid-mediated modulation of pro-inflammatory pathways. a GR can drive the expression of TLR2 by interacting with STAT5 and NF-κB. TLR2 recognizes the bacterial cell wall component peptidoglycan. Upon stimulation, MYD88 binds to the cytoplasmic portion of TLR2. This event leads to recruitment and activation of IRAK1, IRAK4 and TRAF6, ultimately leading to the downstream signaling activation of pro-inflammatory transcription factors which drives the expression of inflammatory cytokines, including IL-6 and IL-8. b, c Glucocorticoids regulate the expression of NLRP3 and P2Y2R by mechanisms that are not well understood. NLRP3 regulates the immune system response to injury or pathogens by sensitizing macrophages to extracellular ATP (danger signal) and inducing the synthesis of IL-1β. P2Y2R is a G-protein-coupled receptor that is activated in response to ATP, which stimulates the activation of PLC (phospholipase C) and PKC (protein kinase C), and the subsequent downstream signaling. Glucocorticoids can upregulate the expression of P2Y2R by enhancing the ATP-dependent activation of p38, which leads to the expression of the pro-inflammatory cytokine IL-6.
TNF-α and dexamethasone drive the binding of NF-κB and STAT to their respective consensus sequences in the TLR2 promoter, leading to TLR2 induction. Furthermore, chromatin immunoprecipitation (ChIP) assays showed that GR is recruited to the TLR2 proximal promoter region in response to dexamethasone, which perhaps contributes to GR-TNF-α effects on TLR2 expression [72]. Although these results appear paradoxical, studies in TLR2 knockout mice have shown that TLR2 deficiency is associated with reduced corticosterone levels, morphological alterations in adrenocortical tissue and impairments in the activation of the inflammatory response following LPS administration [74]. Therefore, these results support the existence of positive feedback between glucocorticoid secretion and the activation of the TLR signaling pathway.
Interestingly, genome-wide microarray studies in A549 cells treated with dexamethasone and TNF-α showed that more than 800 genes were co-regulated significantly by dexamethasone and TNF-α, including the acute-phase protein SerpinA3 secreted into circulation during acute and chronic inflammation [75]. In vitro and in vivo co-administration of glucocorticoids and TNF-α resulted in synergetic increases of SerpinA3 mRNA and protein levels. In addition, ChIP assays indicate that GR binding at the SerpinA3 transcription start site is more robust when cells are co-treated with dexamethasone and TNF-α [75]. These data revealed a novel signaling pathway by which glucocorticoids exert pro-inflammatory actions via interactions with inflammatory cytokines. These results provide a potential explanation for some of the negative side effects of long-term glucocorticoid treatment.
Genome-wide ChIP-chip assays showed that treatment of AtT-20 (pituitary adenoma cell line) cells with leukemia inhibitory factor (LIF), a member of the IL-6 family, and/or dexamethasone potentiates STAT3 and GR recruitment to many STAT3 target genes [76]. LIF signaling on its own was found to modulate a very limited gene subset while dexamethasone co-administrated with LIF leads to a significant upregulation in the expression of genes involved in the cellular defense response, including genes involved in the innate immune response and in the hepatic acute-phase response [76]. Interestingly, similar results were observed in vivo in response to LIF-dexamethasone co-treatment [76]. Therefore, glucocorticoids can work synergistically with pro-inflammatory mediators to reinforce the defense mechanisms to ensure clearance and removal of pathogens [14,76].
Glucocorticoids have also been shown to induce the expression of the NOD-like receptor family, pyrin domain containing 3 gene (NLRP3), a central component of the inflammasome, in both cultured and primary macrophages (fig. 5b) [77]. Glucocorticoid induction of NLRP3 sensitized macrophages to extracellular ATP, which resulted in the secretion of pro-inflammatory cytokines, IL-1β, TNF-α and IL-6 [77]. In addition, glucocorticoids have been reported to induce the expression of the purinergic receptor P2Y2R (fig. 5c). Activation of P2Y2R enhances IL-6 secretion by endothelial cells in response to ATP [78]. Thus, glucocorticoid-mediated activation of TLR2, NLRP3, P2Y2R and potentiation of TNF-α- and LIF-regulated pro-inflammatory genes provide a potential mechanism by which these hormones exert pro-inflammatory actions in response to stress [14].
Concluding Remarks
The discovery that glucocorticoids, the most widely prescribed drugs for the treatment of inflammatory disorders, can exert both pro-and anti-inflammatory actions suggests that the effects of glucocorticoids are more complex than previously recognized. The mechanism(s) by which the same protein can regulate these two opposing processes is only beginning to be understood.
The positive regulation of components of the innate immune response suggests that glucocorticoids prepare the immune system for a quick and efficient response to pathogens [14]. Thus, glucocorticoid-mediated initial pro-inflammatory activity is perhaps required for a functional immune system. Studies on T cell receptor-deficient mice have shown that GR expression is essential for survival during polyclonal T cell activation. GR modulates the activation of both Th1 and Th2 cells, and limits the severity of the inflammatory process by regulating T cell expression of pro-inflammatory molecules [79]. In humans, glucocorticoid deficiency is commonly associated with a defective immune response and recurrent infections [80]. In addition, studies on adrenalectomized rats showed that complete removal of glucocorticoids leads to an increase in susceptibility to LPS-induced inflammation [81]. These findings suggest that glucocorticoids play an important role in priming the immune system to respond to injury, and controlling the release of inflammatory molecules to prevent an overreaction.
In summary, the nature of the response to glucocorticoids relies on several factors, including the duration of the stimulus (acute or chronic) and the physiological state of the immune system. In pathological situations, glucocorticoids may function as anti-inflammatory molecules to control the process. In contrast, under normal physiological conditions, glucocorticoids may play a pro-inflammatory role.
Future studies are needed to characterize the tissue-specific effects of the pro- and anti-inflammatory roles of GR signaling. Understanding of the physiological effects of glucocorticoids should enable us to decipher the mechanisms governing GR actions, and ultimately to develop therapeutic strategies to take advantage of the capacity of GR to elicit pro-and anti-inflammatory effects.
Acknowledgments
We would like to apologize to those colleagues whose work we were unable to cite owing to space limitations. We would like to thank these individuals for all their work that has significantly contributed to our understanding of GR molecular actions. We also would like to thank Dr. Mahita Kadmiel, Dr. Sivapriya Ramamoorthy, Dr. Shannon Whirledge and Alyson Scoltock for their helpful input. Finally, we would like to acknowledge NIEHS arts and photography for their assistance. This work was supported by the NIEHS Intramural Research Program of the National Institutes of Health.
