

Abstract
Alzheimer's disease (AD) is the most prevalent form of dementia which affects people older than 60 years of age. In AD, the dysregulation of the amyloid-beta (Aβ) level leads to the appearance of senile plaques which contain Aβ depositions. Aβ is a complex biological molecule which interacts with many types of receptors and/or forms insoluble assemblies and, eventually, its nonphysiological depositions alternate with the normal neuronal conditions. In this situation, AD signs appear and the patients experience marked cognitional disabilities. In general, intellect, social skills, personality, and memory are influenced by this disease and, in the long run, it leads to a reduction in quality of life and life expectancy. Due to the pivotal role of Aβ in the pathobiology of AD, a great deal of effort has been made to reveal its exact role in neuronal dysfunctions and to finding efficacious therapeutic strategies against its adverse neuronal outcomes. Hence, the determination of its different molecular assemblies and the mechanisms underlying its pathological effects are of interest. In the present paper, some of the well-established structural forms of Aβ, its interactions with various receptors and possible molecular and cellular mechanisms underlying its neurotoxicity are discussed. In addition, several Aβ-based rodent models of AD are reviewed.
Introduction
Alzheimer's disease (AD) was first described by the German psychiatrist, Alois Alzheimer, in the early 1900s [1] and is now considered the most prevalent progressive neurodegenerative disorder, responsible for 75% of all dementia cases [2,3]. It affects approximately 35.6 million people worldwide. This will increase with population aging [4] and will probably affect nearly 106.8 million people by 2050 [5]. It causes mental and cognitive deficits such as impaired memory, intellect and personality disorder in people older than 65 years of age [6,7]. In the advanced stages of the disease central sensory procedures, including the visual system, get affected, too [8]. Collectively, AD-associated problems decrease life expectancy, reduce quality of life, cause physical disability [3], and eventually lead to serious problems in daily life activities such as social and occupational functions [9]. To reduce the social and economic costs and the burden of the disease on patients and their families, some remarkable efforts have lately been made to find diagnostic markers which predict the disease earlier [5]. Neuroimaging methods such as magnetic resonance imaging and positron emission tomography have been developed to enable researchers to diagnose AD in its early stages [10,11]. Also, several biomarkers, which are crucial in detecting pathological features of AD, have been found in cerebrospinal fluid (CSF) and can be assessed [12]. From a histological viewpoint, the progression of AD is associated with 3 cardinal neuropathological features: the accumulation of extracellular senile plaques which is mediated by amyloid-beta (Aβ), intracellular neurofibrillary tangles (NFT) and synaptic degeneration (fig. 1) [13,14]. These events mainly occur in the neocortex, hippocampus, and other subcortical regions which are necessary for cognitive function [15]. The appearance of these markers apparently occurs many years prior to the clinical signs and symptoms of the disease, hence they could be good markers for AD prediction [5]. Meanwhile, Aβ peptide is an important risk factor and has a central role in the onset and progression of AD [16]. Aβ is produced in normal individuals but, under certain circumstances, this molecule may aggregate and start disease progression. There is a large body of evidence emphasizing that Aβ oligomers play the main role in neuronal dysfunction and AD [17,18]. In this article, the points that link AD to various aspects of Aβ pathoneurobiology are reviewed, which may help us understand the process of the disease more clearly.

The 3 major features of AD within the neuronal system. a Cleavage of APP, and formation and accumulation of extraneuronal Aβ. b Formation and deposition of intraneuronal NFTs. c Synaptic dysfunction due to Aβ accumulation and its interaction with receptors. AICD = APP intracellular domain.
The 3 major features of AD within the neuronal system. a Cleavage of APP, and formation and accumulation of extraneuronal Aβ. b Formation and deposition of intraneuronal NFTs. c Synaptic dysfunction due to Aβ accumulation and its interaction with receptors. AICD = APP intracellular domain.
Generation and Clearance of Aβ
Amyloid precursor protein (APP) is a single-pass transmembrane protein which is expressed at high levels in the brain and metabolized in a rapid and highly complex fashion [19]. The APP is cleaved by two pathways. In the nonamyloidogenic pathway, the full-length APP is cleaved by α- and γ-secretases. Cleavage via the β- and γ-secretases can be promiscuous and produces several species of Aβ fragments.
β-Site APP-cleaving enzyme 1 (BACE1) is the major β-secretase in the brain [20]. Neurotoxic forms of Aβ created by cleavage of APP initially by BACE1 produce the C99 fragment and soluble APPβ, and the C99 is then cleaved by γ-secretase to produce Aβ (fig. 1a) [21,22,23]. Moreover, both presenilin 1 (PSEN1) and 2 (PSEN2) regulate the proteolytic function of γ-secretase, and mutations in these proteins can change the activity of γ-secretase and increase the ratio of Aβ in early-onset forms of AD [24].
It has also been suggested that increased levels of free cholesterol in neuronal cell membranes may provoke Aβ formation [25]. Both clinical and genetic data emphasize the unique role of Aβ in the pathogenesis of AD [26].
Based on etiological, pathological, genetic, and biochemical aspects of the disease, AD is divided into two major forms: familial and sporadic [6,27]. It has been known that mutations within the APP gene result in the appearance of familial-type (early-onset autosomal-dominant) AD. On the other hand, the existence of an extra copy of the APP gene, as in patients with Down's syndrome, predominantly leads to the development of AD in the fifth decade of life [28]. In young brains and under normal conditions, there is an equilibrium between the production and elimination of Aβ that maintains Aβ at constant levels, which is known as steady state [29]. However, in aging and pathological conditions such as metabolic disorders and excitotoxicity, the formation and clearance of Aβ have disturbances [21] that lead to an accumulation of Aβ and senile plaque formation [3].
The imbalance of the Aβ level in AD may be due to its production and clearance in the brain. There are different pathways such as the activation of degrading enzymes, receptor-mediated cellular and vascular clearance and other mechanisms by which Aβ is cleared in the brain [30,31,32,33]. Some receptors such as low-density lipoprotein receptor-related protein 1 (LRP1) play an important role in the receptor-mediated clearance of Aβ [30]. Studies have shown that conditional knockout of LRP1 in mouse forebrain neurons increased brain Aβ levels and exacerbated amyloid plaque deposition selectively in the cortex [30]. Also, P-glycoprotein has been suggested to be involved in Aβ clearance as an Aβ efflux pump at the blood-brain barrier (BBB) [31]. Experimental studies have shown that the ablation of P-glycoprotein at the BBB enhanced Aβ deposition in the brain of an AD mouse model [32].
Furthermore, Aβ is degraded by several peptidases, principally two zinc metalloendopeptidases referred to as neprilysin and insulin-degrading enzyme [33]. Studies have shown that neprilysin knockout mice have increased levels of Aβ peptides in the brain. Moreover, the activity of neprilysin is reduced in the cortex and hippocampus of AD patients [34]. Insulin-degrading enzyme is another regulator of Aβ levels in neuronal cells. Genetic studies have shown that insulin-degrading enzyme gene variations are associated with the clinical symptoms of AD [35]. Endothelin-converting enzyme is another degrading enzyme which is expressed in neural tissues, cleaves ‘big endothelin' to produce the vasoconstrictor endothelin-1 and has a principal role in the degradation of Aβ [36].
Population studies have demonstrated that apolipoprotein E (ApoE) ε4 allele is a strong risk factor for late-onset AD [27]. ApoE, the dominant cholesterol and lipid carrier in the brain, is critical for Aβ catabolism. Also, ApoE receptors have been implicated in the clearance of Aβ across the BBB in AD [37]. Impaired clearance of Aβ may also cause sporadic AD through interactions with ApoE4, decreased catabolism of Aβ via reduced proteolytic enzymes, impaired transport across the BBB, or impaired CSF transport [38].
Concentration-Related Behavior of Aβ
Interestingly, it should be pointed out that a very low amount of Aβ may have a role in neural development [39] and in the regulation of cholinergic neurotransmission [21]. It has been demonstrated that Aβ1-40 at a nanomolar concentration inhibits the oxidation of CSF and plasma lipoproteins [40]. Besides, neurons in response to oxidative conditions overexpress Aβ to attenuate oxidative stress outcomes. The in vitro evaluations of the antioxidant activity of Aβ have demonstrated that it is able to protect neurons from neurotoxicity in a concentration-dependent manner [28]. Aβ that accumulates along cerebral blood vessels is known as cerebral amyloid angiopathy. This is frequently seen in AD cases and represents one of AD's histopathological hallmarks. [41]. It causes vasoconstriction and dysregulation of vascular tone.
Hence, it appears that Aβ in higher concentrations, besides neurotoxicity, impairs blood flow within the cerebral structure and accelerates neuronal dysfunction.
Structure-Related Toxicity
Aβ assemblies are divided into three distinct groups (based on length, molecular weight, and microscopic dimensions) as follows: (1) very short oligomers, (2) Aβ-derived diffusible ligands and (3) protofibrils. Very short oligomers of Aβ are referred to as dimmer and hexamer forms of Aβ. Aβ-derived diffusible ligands are small oligomers and their molecular weights range from 17 to 42 kDa [26]. Finally, protofibrils are transient structures which appear prior to the formation of mature amyloid fibrils and can be named prefibrillar assemblies [29].
In their aggregated form, these peptides are able to induce neurotoxicity [15]. This ability, along with the appearance of novel structures, disrupts synaptic functions [29]. Aβ in its specific structural forms provokes nitric oxide formation and an influx of calcium ions which might eventually lead to the formation of peroxynitrite radicals and cell death [42].
It is to be noted that the accumulation of soluble oligomers (nonfibrillar), but not of monomers or insoluble assemblies, has the same neurotoxic effects [43,44]. Interestingly, some studies have indicated that soluble forms of Aβ can impair long-term potentiation and avoidance learning in animal models [45]. Moreover, some of the biological events such as the production of new proteins and the generation of dendritic spines which are involved in memory functions are influenced by the soluble form of Aβ [46]. Therefore, neurodegeneration in AD is mediated in part through soluble forms of Aβ. This form of Aβ is increased in the brain of AD patients [44] and is detectable in their CSF and plasma [47]. Some studies have demonstrated that soluble Aβ concentration correlates with cognitive decline in AD-affected individuals [43]. Hence, it may be considered as a reliable predictor of AD [28]. This idea is also supported by animal studies [48,49], in which soluble forms of Aβ levels correlate with AD progress in the transgenic mouse model of AD but plaque numbers or insoluble Aβ levels have no significant relationship with the severity of AD [48]. Based on these findings, some recent studies have shown that Aβ dimers isolated from human brains are the most toxic species of peptide [49,50].
Aβ Interactions with Receptors
Ryanodine receptors are expressed in the soma, proximal dendrites, and distal processes and spines of neuronal cells [51]. It has been shown that Aβ increases the expression and activity of ryanodine receptor 3 and subsequently these receptors disrupt intracellular Ca2+ levels. The disruption of intracellular Ca2+ homeostasis may have a role in AD pathology [52].
Also, N-methyl-D-aspartate (NMDA) receptor stimulation and intracellular signaling of AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) receptor trafficking are influenced by the soluble form of Aβ [46]. Aβ interacts with NMDA receptors, which are responsible for maintaining glutamate homeostasis within the neurons. Dysregulation of this hemostasis may lead to excitotoxicity and impacts on the neuronal plasticity [53].
LRP1 is a transmembrane protein which acts as a signaling receptor and cargo transporter. It has some essential neural functions, including the process of APP and modulation of the toxicity of the resulting Aβ. Furthermore, LPR1 modulates NMDA receptor function [54]. LRP1 and its ligands, ApoE and α2-macroglobulin, are involved in Aβ deposition through sequestration and removal of its soluble forms [55].
Sorting protein-related receptor (sorLA) is another low-density lipoprotein receptor family which is expressed in neurons and controls APP trafficking/processing and regulates its conversion to Aβ [56]. AD progression disrupts the expression of sorLA and its regulatory function [57]. sorLA gene-inherited variants may regulate the tissue-specific expression of sorLA, which may be associated with late-onset AD [58].
CD36 is an innate immunity receptor which is present in endothelial cells and microglia. It binds to Aβ and activates reactive oxygen species production, vasoconstriction, and vascular tone dysregulation. Collectively, these events may provoke neuronal damage in disease progress [59].
N-formyl peptide receptor like-1 (FPRL-1) is a seven-transmembrane, G-protein-coupled receptor. It is expressed on human mononuclear monocytes like microglia. Aβ1-42 is a chemotactic agonist for this receptor. Following infiltrating senile plaques in the brain of AD patients, its expression is increased in inflammatory cells. Thus, this receptor may have a role in inflammation seen in AD [60].
Moreover, Aβ interacts with other receptors such as tyrosine kinase (TrkA), pan-neurotrophin p75 (p75NTR) and α7 nicotinic acetylcholine (nAChR) [61].
Several neurotrophin signaling pathways may be activated in response to Aβ. Aβ increases the nerve growth factor and its receptor TrkA expression [62]. In addition, the interaction of Aβ with p75NTR has a pivotal role in AD pathogenesis. Cholinergic basal forebrain neurons express p75NTR receptors and Aβ may induce apoptosis through these receptors [61]. Moreover, TrkA reduces β-cleavage of APP, whereas p75NTR activates this process. Also, normal aging activates Aβ generation in the brain by ‘switching' from the TrkA to the p75NTR receptor system [62].
The interaction of Aβ with α7 nAChR promotes the endocytosis of NMDA receptors and impairs normal cholinergic neurotransmission [29]. Furthermore, Aβ shows antagonistic effects on α7 nAChR in a dose-dependent manner, and its pathological function may partially correlate with the blocking of these receptors [39].
Aβ and the Immune System
In response to Aβ-induced neurotoxicity in AD, both humoral and cellular immunity are activated. Aging is commonly accompanied by a progressive dysregulation of the immune response, mainly due to alterations of cellular immunity. In comparison to these changes in cellular immunity, many features of innate immunity are relatively well maintained with age [63].
A number of publications have reported the presence of anti-Aβ antibodies in the blood and CSF of patients with AD and healthy subjects [64]. Also, some drugs have recently been designed based on fully human anti-Aβ monoclonal antibodies for the clearance of Aβ [65,66].
Aβ deposition is responsible for microglia activation. Aβ contributes to the enhancement of the inflammatory response by NF-κB stimulation, a nuclear factor that is implicated in cytokine production and also regulates the ERK (extracellular signal-regulated kinase) and MAPK (mitogen-activated protein kinase) pathways that lead to cytokine and chemokine production [67]. Toll-like receptors (TLR) are important for regulating microglial responses to Aβ and fibrillar Aβ triggers microglia inflammatory cytokine production via TLR4-TLR6 heterodimers, whose assembly is regulated by CD36. Modification of the inflammatory state of microglia/macrophages may have an axial role in AD-related pathology [68].
Prion-Like Mechanism of Aβ Toxicity
The concept of ‘prion-like' has been suggested to explain the pathogenic mechanism of all the principal neurodegenerative disorders associated with protein misfolding, including AD [69].
Like prions, Aβ peptides may fold in different manners, thus giving rise to ‘strains' with specific pathological aspects. Also, Aβ oligomers show a high affinity for binding to cellular prion protein (PrPC). Aβ oligomers alter the activity of the Src family tyrosine kinase Fyn which participates in the PrPC-regulated signaling pathway [70]. Also, Aβ binds to postsynaptic PrPC and activates Fyn to impair neural function [71]. It has been reported that NMDA receptors are necessary for the transduction of Aβ toxic signaling [70]. Another study has indicated that the Aβ-PrPC complex exerts its cytotoxicity via transmembrane LRP1 [72]. Additionally, the pattern of transmission and the spreading of Aβ are similar to prions in the brain of AD patients [73].
Aβ and Neurofibrillary Tangles
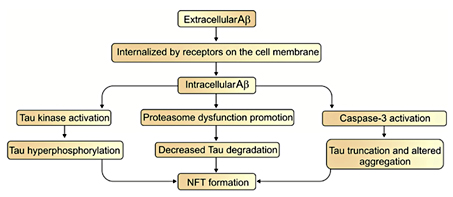
Tau is a microtubule-associated neuronal protein. It is generated by neurons and is localized in the cell body and axons [74,75]. Under normal conditions, nerve growth factor increases tau expression during neuronal development [76]. However, in some pathological conditions it is also produced by glial cells [75,77]. Although tau's main expression region is in the central nervous system, its mRNA is found in peripheral tissues, too [75]. In the brain, six tau isoforms are generated from a single gene through the alternative splicing of mRNA [74,77]. Under normal conditions, tau promotes the assembly of tubulin into microtubules and maintains their stability [78,79]. Besides this function, it interacts with spectrin and actin filaments [75] and has a role in TrkA receptor-mediated signal transduction [74]. It is thought that tau abnormalities result in NFT production and neuronal death that eventually leads to dementia [80]. Accumulations of tau occur in a wide spectrum of neurodegenerative disorders such as progressive supranuclear palsy, corticobasal degeneration, Pick's disease, argyrophilic grain disease, the Parkinson-dementia complex of Guam, and AD [77]. The hyperphosphorylated form of tau protein exerts neurotoxic effects in these diseases which are collectively termed as tauopathies [74]. Tau in its hyperphosphorylated forms accumulates in somatodendritic parts of the neurons and becomes the core component of NFTs [76]. NFTs are structurally paired helical filaments which are composed of hyperphosphorylated tau proteins and neurofilaments [15,77]. The formation of NFTs is directly associated with neuronal dysfunction and the number of NFTs are related to the degree of dementia in AD [81]. It has been suggested that many years prior to the appearance of clinical signs of AD [5] the deposition of both NFT and Aβ occurs within the neocortex, hippocampus, and other cognition-related subcortical structures [15]. Increasing evidence shows that Aβ can be internalized or produced inside of the cells. This provides the opportunity for Aβ to facilitate NFT formation [82]. On the other hand, the disruption in tau formation can influence the production of Aβ and amyloid plaques [82,83]. Three major pathways have been proposed to illustrate the link between Aβ and tau pathology. First, the activation of tau kinases by Aβ induces NFT formation through tau hyperphosphorylation. Second, Aβ decreases tau degradation by the promotion of proteasome dysfunction and, finally, Aβ activates caspase-3, which causes the truncation of tau and altered tau aggregation that leads to NFT formation (fig. 2) [84]. Since immunotherapy with anti-Aβ antibodies in a triple transgenic mouse model of AD reduced Aβ accumulation and slowed the formation of NFT, it seems that Aβ is also involved in the formation of NFTs [85]. This surmise is supported by the fact that during AD development NFTs gradually accumulate in limbic areas and in the isocortex after Aβ aggregation and cause dementia and cognitive dysfunction [80].
Synthetic and Naturally Secreted Aβ
Aβ assemblies are classified as synthetic and naturally secreted forms. Synthetic Aβ oligomers include 10, 40 or 42 peptides and mimic the most common forms of Aβ which are found in both AD and the normal human brain [86]. It has recently been shown that synthetic Aβ dimers which mimic the natural ones can make some stable protofibrils that persist for a long time and impair synaptic plasticity [87]. Moreover, these synthetic forms of Aβ are chemically defined and can be readily synthesized and biophysically characterized [46]. On the other hand, for the achievement of adequate memory disruption in animals these forms of Aβ must be used at higher doses [50].
Naturally secreted Aβ peptides are usually obtained from CHO (cultured Chinese hamster ovary) cells which stably overexpress APPV717F (a mutant form of APP known to cause the familial form of AD) [50,88] and release soluble oligomers (dimmers, trimmers and tetramers) and monomers of Aβ [50,86]. Importantly, biochemical properties of Aβ assemblies derived from this cell line have been reported [50]. There are some other natural forms of Aβ which can be produced in the brains of patients with AD, including Aβ25-35. Aβ25-35 is a fragment of full-peptide Aβ [89]. It forms β-sheets similarly, which provoke neuronal cell death, memory impairment and synaptic damage [90,91]. Recent evidence shows that a single-dose intracerebroventricular injection of this form can mimic some main neuropathological signs of AD in its early stages in rats [92].
As mentioned above, synthetic forms of Aβ contain predefined lengths of peptides, whereas CHO-derived Aβ assemblies, like other naturally produced brain and CSF Aβ peptides, are heterogeneous in length. On the other hand, unlike the synthetic forms of Aβ, naturally produced Aβ peptides exert their biological effects only at very low doses and, therefore, they are able to effectively disrupt memory and long-term potentiation in animal models [48,50].
Aβ-Based Rodent Models
Aβ is able to induce neurotoxicity directly and cause destruction in cholinergic basal forebrain projections [21]; moreover, it can disrupt memory skills in experimental models. Different doses of synthetic and/or naturally secreted Aβ peptides are stereotactically delivered into the brain through intracerebroventricular [46,48,93,94] and/or intrahippocampal injections for the induction of experimental AD in rodents [95].
Nowadays, transgenic and knockout rodents [96,97] have been developed as well as other transgenic models such as Caenorhabditis elegans [98,99], Drosophila melanogaster [100] and viral vector-driven models of AD [101], but mice are by far the most used genus. Due to the complexity of AD, it has been difficult to create a transgenic model that replicates the multiple characteristics of the disease. Thus, most of the transgenic models that have been generated to date exhibit some of the major pathological hallmarks of AD [102]. By considering amyloid importance in AD pathogenesis, the main focus of modeling studies is on amyloid deposition [101]. After the discovery of familial AD mutations in APP, modeling studies focused on making AD models based on the overexpression of transgenes containing familial AD mutations [103]. The first predication of the AD transgenic model was reported in 1995 [104], which expressed high levels of mutant APP to generate extracellular Aβ in specific parts of the brain [105]. Consequently, numerous models have been successfully developed which are deposits of amyloid plaque [106]. Some of these models are shown in table 1.

The type of mutation affects plaque formation in models. For example, mutations at the N terminus of Aβ lead to an increase in Aβ40 and Aβ42, whereas mutations at the C terminus lead to an increase in the Aβ1-42 form [102]. However, a mutant of the amyloid-related gene carrier has been crossed with the human APP overexpressing line in these models. Most of these models express transgenic APP at an extremely higher level than the endogenous APP. Most of the transgenic animals exhibit age-dependent amyloid deposition similar to that found in AD.
Conclusion
The most common neurodegenerative disorder and the most important cause of dementia in elderly people appears to be AD, and Aβ peptide has a substantial role in its pathogenesis. The appearance of Aβ occurs many years before the clinical signs and symptoms of the disease, so it could be a reliable biomarker for AD prediction. As indicated, Aβ plays an important role in the formation of both amyloid plaques and NFTs, which gradually leads to AD. Aβ deposition leads to synaptic degeneration and interacts with different types of central nervous system receptors; hence, it disrupts neuronal homeostasis. Moreover, Aβ deposition along the cerebral vessels alters their tonicity and triggers some of the cerebrovascular deficits. Furthermore, its accumulation disrupts intracellular Ca2+ homeostasis which ultimately reduces neuronal Ca2+ buffering capacity and increases excitotoxicity outcomes. Also, Aβ peptides may fold in different ways and show a prion-like pathology in the brain of AD patients.
Recently, most of the efforts have been directed to controlling the production and clearance of Aβ. Interestingly, given this, anti-Aβ monoclonal antibodies have been developed as a novel strategy in the treatment of AD. Furthermore, animal models related to Aβ neurotoxicity have been developed for the better understanding of AD and for the testing of new strategies against it. The exact mechanisms of this peptide are unclear and more studies need to be done to clarify them.
Acknowledgment
The authors would like to express their gratitude to Dr. Mohammad R. Rashidi, Vice Chancellor, Research and Technology, Tabriz University of Medical Sciences, Tabriz, Iran for spiritual auspices.
Disclosure Statement
The authors have no conflicts of interest to disclose.

