

Abstract
The anti-diabetic and oral hypoglycaemic agent metformin, first used clinically in 1958, is today the first choice or ‘gold standard' drug for the treatment of type 2 diabetes and polycystic ovary disease. Of particular importance for the treatment of diabetes, metformin affords protection against diabetes-induced vascular disease. In addition, retrospective analyses suggest that treatment with metformin provides therapeutic benefits to patients with several forms of cancer. Despite almost 60 years of clinical use, the precise cellular mode(s) of action of metformin remains controversial. A direct or indirect role of adenosine monophosphate (AMP)-activated protein kinase (AMPK), the fuel gauge of the cell, has been inferred in many studies, with evidence that activation of AMPK may result from a mild inhibitory effect of metformin on mitochondrial complex 1, which in turn would raise AMP and activate AMPK. Discrepancies, however, between the concentrations of metformin used in in vitro studies versus therapeutic levels suggest that caution should be applied before extending inferences derived from cell-based studies to therapeutic benefits seen in patients. Conceivably, the effects, or some of them, may be at least partially independent of AMPK and/or mitochondrial respiration and reflect a direct effect of either metformin or a minor and, as yet, unidentified putative metabolite of metformin on a target protein(s)/signalling cascade. In this review, we critically evaluate the data from studies that have investigated the pharmacokinetic properties and the cellular and clinical basis for the oral hypoglycaemic, insulin-sensitising and vascular protective effects of metformin.
Introduction
In his 1957 publication, Jean Sterne [1] was the first to recognise the oral hypoglycaemic efficacy in humans of the dimethylbiguanide, metformin, even though the therapeutic potential of guanidines had been documented much earlier in 1918 by Watanabe [2], who described the effects of subcutaneous injections of guanidine hydrochloride on blood glucose levels in rabbits. Watanabe's studies were influenced by the knowledge that French lilac (Galega officinalis), also known as goat's rue, Italian fitch or professor weed, contains alkaloids such as galegine, which is chemically related to guanidine. French lilac has been used since the Middle Ages for the treatment of polyuria associated with diabetes [3]. This folklore knowledge led to the study of guanidines, which turned out to be toxic for use in humans and also potentially fatal when eaten by goats and sheep, hence the name goat's rue [3]. Thus, synthetic biguanides, such as buformin, phenformin and metformin, were developed as potential therapeutic agents for the treatment of diabetes. Subsequent to the report by Sterne [1], metformin was introduced into the UK in 1958 for the treatment of type 2 diabetes (T2DM) with the trade name Glucaphage®, i.e. ‘glucose eater'. It was not until much later that metformin was approved by the FDA for use in the USA [4]. The long delay could be linked to the withdrawal in 1978 of another biguanide, phenformin, due to its link to a high incidence of lactic acidosis and cardiovascular side effects [5]. Today, metformin is the ‘gold standard' and drug of choice to treat patients with T2DM. It is also prescribed for the treatment of polycystic ovary disease and is being investigated for use as an adjunct agent, together with insulin, for patients with type 1 diabetes (T1DM) [6]. A retrospective analysis has also suggested that metformin may have beneficial effects for patients with cancer [7]. The protective action of metformin against cancer may be secondary to its negative effects on metabolism, thus offsetting the anti-senescence effects of the adenosine triphosphate (ATP)-generating glycolytic ‘Warburg effect' on stimulating tumour growth. Alternatively, the effects of metformin on cancer may be independent of its effects on metabolism and directly related to an inhibition of tumour growth [8,9].
Diabetes mellitus is a major health problem that affects in excess of 350 million people worldwide [10]. There is an increase in the incidence of diabetes worldwide, and, in the case of T2DM, this can be linked to the sedentary lifestyle patterns that also contribute to obesity. There is a particularly high prevalence in the Gulf State countries and, for instance, a cross-sectional study in Qatar that was published in 2009 found that the prevalence of diagnosed and undiagnosed diabetes mellitus among adults was as high as 16.7% [11]. Three years later, a prevalence of 23% was reported [12]. Similar concerns over the rising tide of obesity in children have been expressed in Kuwait [13], and the dramatic increases in the prevalence of obesity and diabetes in the entire Middle East North Africa population are causes for considerable concern [14].
There are three main types of diabetes. (1) Gestational diabetes appears in late pregnancy in about 3-5% of women and usually disappears at parturition. However, the risk of developing T2DM is higher in women who have had gestational diabetes and, furthermore, the incidence is increasing [15]. (2) T1DM is an autoimmune disease that results in the loss of pancreatic beta-cell function and hence the loss of insulin production, and accounts for about 5-10% of the population diagnosed with diabetes. Over the past 30 years, the incidence of T1DM has increased worldwide, suggesting a potential multifactorial environmental link [16]. (3) T2DM is the most common form of diabetes, accounting for >90% of the cases which result from the development of insulin resistance (IR), thus resulting in the reduced ability of tissues in the body to take up glucose. T2DM, although once associated with adults and hence the older terminology of ‘adult onset diabetes', is now frequently also seen in adolescents and children, and the incidence has dramatically increased worldwide [17]. Dabelea et al. [18 ]reported an increase in the incidence in the USA for the period 2001-2009 of 21 and 30%, respectively, for T1DM and T2DM in children and adolescents.
The common feature of gestational diabetes, T1DM and T2DM is the loss of glycaemic control, with the result that, dependent on how effective the therapy is, the patient experiences periods of hyperglycaemia, particularly after meals. The results of 2 large clinical trials, the DCCT (Diabetes Control and Complications Trial), conducted between 1983 and 1993 for T1DM, and the UKPDS (UK Prospective Diabetes Study), conducted between 1977 and 1991 for T2DM, concluded that the tight control of blood glucose levels reduced the incidence of microvascular complications [19,20]. Subsequent studies, such as the EDIC (Epidemiology of Diabetes Interventions and Complications), a follow-up to the DCCT, reported that tight control of blood glucose also reduced cardiovascular morbidity and mortality [21]. The evidence from the UKPDS as well as more recent clinical data assessing endothelial function in patients with T2DM indicates that metformin, in addition to its established actions as an oral hypoglycaemic drug, protects the vascular system from the development of microvascular disease [20,22,23,24].
Diabetes and Vascular Disease
Diabetes mellitus is a multi-systemic disease and can cause multiple organ damage through several mechanisms. Vascular disease is the main contributor to the morbidity and mortality associated with diabetic patients [25]. Endothelial cells are glycolytic, but in the presence of hyperglycaemia, the excess glucose can be shunted into other pathways, such as the polyol pathway or non-enzymatic generation of advanced glycated end-products (AGEs) [26,27]. Oxidative stress is argued to be the primary trigger for the initiation of vascular disease [28]. In the presence of hyperglycaemia, mitochondria-derived reactive oxygen species (ROS) increase, and this increase is believed to be the key trigger for the development of endothelial dysfunction and vascular disease [25]. Changes in endothelial function, often measured as a reduction in endothelium-dependent vasodilatation (EDV) and/or biomarkers such as sICAM-1 (soluble intercellular adhesion molecule-1) and sVCAM-1 (soluble vascular adhesion molecule-1), are considered to be early indicators of the development of cardiovascular disease [29,30,31]. As discussed in the following section, there is extensive literature indicating that mitochondria are a target for the guanidines phenformin and metformin. At an appropriately high local concentration, guanidines and biguanides are inhibitors of mitochondrial complex 1, thus reducing NADH:ubiquinone oxidoreductase activity, reducing cellular metabolism and producing a ‘mild' inhibitory action of the mitochondria respiratory chain. Inhibition of complex 1 has also been advanced as the explanation for metformin's putative, protective effect on the vasculature and its actions to suppress hepatic gluconeogenesis [31,32]. The inhibition of complex 1 reduces the ATP/adenosine monophosphate (AMP) ratio, which, in turn, activates AMP-activated protein kinase (AMPK). However, whether AMPK mediates some/any of the cellular actions of metformin continues to be debated. Of course, negative effects on mitochondrial function could equally contribute to the toxicity of the biguanides. For instance, 24-hour exposure of MIN6 (a pancreatic beta-cell line derived from a mouse insulinoma) to 2 mM of metformin results in the inhibition of mitochondrial complex 1, AMPK activation and cell death; however, beta-cell toxicity is not associated with the therapeutic use of metformin in diabetic patients [33]. Although species and potential tissue-dependent differences may contribute to the differences in tolerance and toxicity of metformin, it is important to fully understand the pharmacokinetic and pharmacodynamic properties of metformin.
The Pharmacokinetics of Metformin
Although the biguanide phenformin was withdrawn in 1978 due to a close association with lactic acidosis and cardiovascular problems, retrospective analyses indicate that metformin is safe, with a very low incidence of serious side effects and a minimal risk for triggering lactic acidosis. Nonetheless, its use should be avoided in patients with severe renal and/or liver dysfunction. As for all drug treatments, physicians should be vigilant for the occurrence of rare side effects [34,35,36,37]. It is possible that the high toxicity of phenformin compared to metformin is related to the much greater affinity of phenformin (>50) for mitochondrial membranes and subsequent suppression of oxidative phosphorylation, although other mechanisms may also contribute [38,39].
To better understand the sites and cellular mechanisms of action, it is necessary to review the pharmacokinetic properties of metformin. Metformin is a biguanide that has a strong base with a pKa of 12.4, and therefore exists predominantly as a protonated cation at physiological pH. Despite its hydrophilicity, metformin can be transported across cell membranes via organic cation transporters (OCT). OCT1 is expressed in the intestine, liver and erythrocytes (among other cell types), OCT2 is expressed in the kidney and OCT3 is the plasma membrane monoamine transporter [40,41].
Metformin is orally administered in the dose range of 500 mg/b.i.d. or t.i.d. and up to a total of 2,550 mg/day or approximately 35 mg/kg/day. Metformin is slowly absorbed from the proximal small intestine. Higher doses slow the rate of absorption and reduce the bioavailability [42,43]. In contrast, phenformin is absorbed well following oral administration and this property may also contribute to the greater likelihood of lactic acidosis with phenformin versus metformin. Steady-state plasma concentrations of metformin have been reported to be between 1 and 10 μM, although initially these are likely higher in the hepatic-portal circulation. Metformin does accumulate in the gastrointestinal tissues; this may be a contributing factor to the gastrointestinal-associated side effects. Metformin also accumulates in erythrocytes, serving as a potential reservoir [43,44,45]. Thus, one can estimate that the plasma concentration of metformin, subject to dose and rate of absorption, may vary from 1 to approximately 50 µM, with the latter higher concentration in the hepatic portal vein and liver, as determined by studying 14C-labelled metformin [45,46]. Accumulation in the liver would also help to explain the assumed primary site of action of metformin as an inhibitor of hepatic gluconeogenesis [46]. Metformin does not undergo significant metabolism and is excreted unchanged (>90%) in the urine following tubular secretion via the OCT2 transporter [42,47]. The rate of metformin absorption is slower than its rate of plasma elimination, and therefore intestinal absorption is the rate-limiting step for metformin action [42]. In contrast, phenformin is subjected to metabolism, with about one third of the administered drug excreted in the urine as the hydroxyl derivative. Polymorphisms in OCTs will affect both the absorption and the secretion of metformin [47]. The plasma half-life of metformin is approximately 2-6 h, but other compartments, such as erythrocytes and the gastrointestinal tract and most likely enterocytes, contribute to a slower half-life for elimination of up to 14 h. This really reflects the absorption-limited kinetics of metformin [42]. Thus, as stated by Sirtori et al. [48]: ‘The very brief plasma half-life of metformin makes significant accumulation with a standard t.i.d. regimen unlikely.' Nonetheless, the accumulation of metformin in erythrocytes might be a source of excess lactate production and potential lactic acidosis, particularly when renal excretion is severely impaired [44].
The primary beneficial effect of metformin in the treatment of T2DM is assumed to be secondary to a reduction in hepatic gluconeogenesis and improved glucose transporter 4 (GLUT-4)-mediated transport of glucose into adipose tissue and striated muscle.
Mode(s) of Action of Metformin: Hepatic Gluconeogenesis and GLUT-4-Mediated Glucose Transport
Metformin and AMPK
Reduction in the rate of glucose production by inhibiting hepatic gluconeogenesis is the principal mechanism whereby metformin improves glycaemic control in T2DM [49]. Metformin has been shown to reduce hepatic IR by correcting the activity of two key enzymes, glucose-6-phosphatase (G6pc) and glucokinase [50]. G6pc is the rate-limiting enzyme for the hepatic production of glucose, and concentrations of metformin in excess of 300 and 1 mM, respectively, reduce the expression of G6pc mRNA and G6pc activity [51]. Metformin has also been shown to suppress gene expression of the rate-limiting enzyme for gluconeogenesis, phosphoenolpyruvate carboxykinase, in rat hepatocytes [52]. Metformin-mediated inhibition of hepatic gluconeogenesis has been linked to the activation of AMPK, the cell's ‘fuel gauge' [53]. AMPK is a phylogenetically conserved serine-threonine kinase composed of an α-catalytic subunit and two regulatory subunits, β and γ; however, there are multiple isoforms of these subunits (α1, α2, β1, β2 and γ1-γ3), with tissue-specific expression suggesting multiple functions [54]. Zhou et al. [55 ]reported that metformin-mediated activation of AMPK was associated with suppression of hepatic gluconeogenesis and fatty acid oxidation and in isolated skeletal muscle, metformin enhanced GLUT-4 mediated glucose uptake. Furthermore, the effects of metformin could be mimicked in rat primary hepatocytes by the application of the cell-permeable AMPK activator, AICAR (5-amino-1-β-D-ribofuranosyl-imidazole-4-carboxamide) and inhibited by the AMPK inhibitor, compound C (6-[4-(2-piperidin-1-ylethoxy)phenyl]-3-pyridin-4-ylpyrazolo[1,5-a]pyrimidine), also known as dorsomorphin [55]. Similarly, in diabetic patients, the intravenous injection of AICAR reduces hepatic glucose output [56]. Metformin-mediated suppression of hepatic gluconeogenic genes has also been linked to the activation of AMPK and the subsequent phosphorylation of serine 436 of the transcriptional co-activator cAMP response element-binding protein, CREB, via protein kinase C (PKC)-ι/λ [57]. These conclusions were based on cell culture studies with primary mouse hepatocytes and hepatoma cells exposed to 10 mM of metformin and mice treated with 200 mg/kg/day i.p. In comparison, the maximum therapeutic dosage of metformin in a man weighing 70 kg is approximately 35 mg/kg/day. Of course, species differences in tissue sensitivity to metformin should be considered, although, in the absence of any evidence for the metabolism of metformin, the comparison of dose ranges and plasma concentration remain the most practical means for evaluating the data in the literature.
Phosphorylation of AMPK leads to a cascade of cellular reaction, including the inactivation of acetyl-CoA carboxylase (ACC), the inhibition of ACC lipogenesis and increased fatty acid oxidation. AMPK also decreases the expression of sterol regulatory element-binding protein 1 and genes such as FAS and S14. These changes further decrease lipogenesis and increase fatty acid oxidation, which, in turn, decreases hepatic steatosis and lowers the levels of very-low-density lipoprotein and triglycerides. Metformin-mediated increases in AMPK have also been argued to explain the effects of metformin in enhancing GLUT-4-mediated glucose utilisation in human skeletal muscle [58]. Metformin has been reported to inhibit AMP deaminase, and the resultant increase in AMP activates AMPK, thus providing an explanation for metformin-mediated GLUT-4-facilitated glucose uptake into skeletal muscle [59]. Shaw et al. [60] provided evidence that metformin targets the serine-threonine kinase, liver kinase B1 (LKB1), which is upstream of AMPK, as mice deficient in LKB1 failed to respond to an in vivo treatment with metformin of 250 mg/kg/day, a dose that is approximately 7 times the maximal daily dose for humans. However, the hypothesis that the direct action of metformin on LKB1 and/or AMPK mediates the effects of metformin in the liver has been challenged because a hypoglycaemic response to metformin is still observed in mice that are genetically deficient in AMPKα1 and carry a liver-specific knockout of both AMPKα2 and LKB1 [61]. Conversely, metformin-mediated increases in AMPK-mediated phosphorylation and inhibition of ACC are argued to be the basis for the insulin-sensitising effect of metformin to suppress hepatic lipogenesis and lipid accumulation. These data were generated in primary mouse hepatocytes in the presence of 500 μM of metformin and mice treated either acutely with 200-400 mg/kg or chronically with 50 mg/kg of metformin [62]. Of interest, Hawley et al. [63] reported a slow time-dependent activation of AMPK of up to 72 h in H4IIE rat hepatoma cells with 50 μM of metformin, but faster activation with 250 μM. Conceivably, metformin does not need to maximally activate AMPK to produce a therapeutic effect as, for instance, phosphorylation of the AMPK target protein ACC in H4IIE cells occurs at very low levels of phosphorylated AMPK [63]. Turban et al. [64] have presented a compelling argument that metformin-stimulated glucose uptake into L6 skeletal muscle cells is mediated by an AMPK-independent action involving PKC activation. A high concentration of 1 mM of metformin was used in their study, but no change in AMPK activation was reported. Metformin-stimulated glucose uptake was shown to be sensitive to a non-specific PKC inhibitor, Ro-31-8220, but was unaffected in L6 cells depleted of PKC-α, thus suggesting a role for a non-PKC-α novel/conventional PKC [64]; however, the non-PKC-related effects of Ro-31-8220 may also contribute. Collectively, these data suggest that both AMPK-dependent and AMPK-independent effects of metformin contribute to its hypoglycaemic actions. More studies are required to confirm whether metformin-mediated increases in AMPK are essential for its therapeutic benefits.
Metformin and Mitochondria
Metformin inhibits mitochondria complex 1, NADH: ubiquinone oxidoreductase (or ubiquinone) activity, which is the entry point for mitochondrial oxidative phosphorylation [54,65,66,67]. Unlike another complex 1 inhibitor, rotenone, which decreases ROS generation but can also result in ROS overproduction, metformin only seems to reduce ROS. The difference between rotenone and metformin is that metformin may selectively inhibit reverse electron flux [68]. In a 2014 review article, Pernicova and Korbonits [69] concluded: ‘Metformin acts as a metabolic inhibitor and alters both whole-body and cellular energy metabolism.' It is argued that the hypoglycaemic effects of metformin are secondary to the interaction of metformin with mitochondrial complex 1 and a subsequent lowering of the cellular ATP/AMP ratio. ATP inhibits the binding of AMP to AMPK and a small change in the ATP/AMP ratio would produce a proportionally larger change in AMPK activity [70]. Raising AMP levels would lead to the activation of AMPK, but this does not necessarily imply that AMPK mediates the therapeutic actions of metformin, as AMPK has multiple cellular targets [54].
The argument that the basis of metformin's action as an oral hypoglycaemic drug stems from an action on complex 1 has been championed by El-Mir et al. [65] and Owen et al. [67]. El-Mir et al. [65] reported that the IC50 for metformin as an inhibitor of oxygen uptake in intact, isolated mitochondria was approximately 1 mM. No effect of metformin was observed in permeabilised hepatocytes, suggesting to the authors that metformin's action was mediated by a receptor-activated signalling mechanism. A 30-min exposure to 10 mM of metformin reduced the ATP/ADP ratio in hepatocyte mitochondria by approximately two thirds [65]. Owen et al. [67] provided data that indicated that exposure to 50 and 100 µM of metformin in a time-dependent manner inhibited mitochondrial respiration in permeabilised rat hepatoma cells by 12.6 and 29% and 26 and 37% at 24 and 60 h, respectively. However, comparing the ability of phenformin and metformin to inhibit NADH-dependent respiration in isolated rat mitochondria, phosphoenolpyruvate K0.5 concentrations of of 50 micromolar and 14.9 mM were obtained. In order to preserve mitochondria integrity, these experiments were performed at 8°C, and thus the inhibitory coefficients of the drugs may have been underestimated. The peak plasma concentration of metformin has been reported to approximate 10 µM [43,71]. So the question is: ‘Would you expect metformin to reach mM concentrations in the liver in vivo particularly given the slow absorption kinetics and short plasma t1/2 of metformin?' Based on its known pharmacokinetic properties, the most likely answer is ‘no'. However, although the pharmacokinetic properties of metformin suggest otherwise, it can also be argued that a slight reduction in complex 1 activity could explain the effects of metformin.
Based on thermodynamic considerations extrapolated from data in an earlier study by Davidoff [72], Owen et al. [67] argued that driven by the mitochondrial membrane potential, the positive charge on metformin would promote a 1,000-fold accumulation of metformin in the matrix of mitochondria with a theoretical phosphoenolpyruvate concentration of 80 µM for metformin inhibition of complex 1. Although Davidoff [72] did indeed show a slow uptake of phenformin into mitochondria isolated from the guinea pig that was sensitive to pH changes and correlated with the degree of respiratory inhibition, the equivalent data were not presented for metformin. Phenformin, by a concentration-dependent mechanism (5-50 μM), has also been shown to enhance calcium uptake into mitochondria [73]. The effect of phenformin on calcium transport occurs at concentrations that are >100 times lower than those required to inhibit mitochondrial respiration, leading to speculation that altered mitochondrial calcium homeostasis plays a role in the hypoglycaemic action of phenformin [73]. Furthermore, the uptake of phenformin into mitochondria is inhibited by magnesium, suggesting competition for a cation carrier [74]. The data from the study by Davidoff [72] are suggestive that the distribution of phenformin is determined by the trans-membrane pH gradient and the trans-membrane mitochondrial electrical potential. However, there is no evidence from in vivo data that metformin accumulates to a level necessary to affect mitochondria respiration. In fact, Wilcock et al. [46] demonstrated that the 78% distribution of 14C-metformin in the mouse liver was associated primarily with the cytosolic fraction and <10% with the mitochondria. As already noted, Schäfer [39] reported that metformin has a low binding affinity for mitochondrial membranes whereas that for phenformin is approximately 50 times greater. Thus, although alkyl-substituted biguanides avidly bind to biological membranes, this is not the case for metformin, which has two methyl side chains as opposed to the phenethyl side chain of phenformin, and, therefore, a greatly diminished affinity for the hydrophobic nature of such membranes. In addition, metformin is a strong base and the pH gradient in mitochondria would serve to oppose accumulation, although it should be noted that the membrane potential may promote entry. Furthermore, a therapeutic concentration of metformin, 10 μM, potentiated the anti-gluconeogenic effect of insulin in isolated rat hepatocytes without affecting the ATP content of mitochondria [75]. In contrast, the anti-gluconeogenic action of 10 mM of metformin is associated with an increased mitochondrial NADH:NAD+ ratio, suggesting a change in the redox state and also indicating that the concentration-dependent effects of metformin on mitochondria must be considered when making conclusions about the therapeutic actions of the drug.
Concentration-dependent inhibitory effects of metformin have been reported to be in the range of 250 μM to 2 mM on ATP content in hepatocytes from mice either expressing or deficient in AMPKα1 and α2 subunits or the upstream kinase LKB1 [61]. It has also been reported that ATP levels were decreased in mice treated with 50 mg/kg of oral metformin for 5 days, whereas AMP levels were decreased by doses of 20 or 50 mg/kg for the same time period [61]. Collectively, these data stress the need to seek alternative explanations for the effects of metformin on cell metabolism because the inhibition of complex 1 is only clearly evident at very high concentrations of metformin and therefore seems to be an unlikely target for explaining metformin's therapeutic efficacy as an oral hypoglycaemic agent. This conclusion is substantiated by the report from Larsen et al. [76 ]that biopsies of skeletal muscle from T2DM patients treated with metformin do not show suppressed complex 1, and that inhibition of complex 1 by metformin in rat skeletal muscle is concentration-dependent with a threshold of 1 mM. However, therapeutic concentrations of metformin do normalise mitochondrial hydrogen peroxide generation and this could contribute to a metformin-mediated reduction in the production of ROS [77]. Furthermore, Hawley et al. [63] demonstrated that metformin did not activate (phosphorylate) AMPK in cell-free protocols, but did activate AMPK in CHO (Chinese hamster ovary) and H4IIE rat hepatoma cells without affecting the ATP/AMP ratio. In addition, although the same concentration range of 250-2,500 μM of metformin was required to activate AMPK in both cell types, the CHO cells, but not the H4IIE cells, were resistant to the mitochondria ATP synthase and respiratory chain inhibitors, oligomycin and antimycin A, strongly suggesting that the effects of metformin on AMPK activation are not linked to the inhibition of complex 1.
Metformin and the Inhibition of Adenylate Cyclase
An increase in AMP will also result in P-site-mediated inhibition of adenylate cyclase [78]. Thus, if metformin reduces the ATP/AMP ratio in the liver, depression of glucose output would also result from a reduction in glucagon receptor signalling and the Gs-coupled activation of adenylyl cyclase/cAMP/PKA-mediated increases in liver glycogenolysis and gluconeogenesis. This hypothesis has been advanced by Miller et al. [79], with supportive data obtained from mouse primary hepatocytes treated with either phenformin or metformin and mice treated via gavage in vivo with 500 mg/kg of metformin, which is approximately 15 times the maximal daily dose used for humans. While the data are supportive of the hypothesis, the comparatively high concentrations of metformin that are required to reduce cAMP levels (an IC50 of approx. 200 μM) suggest caution in relating the inhibition of complex 1 to the therapeutic efficacy of metformin as a hypoglycaemic agent. In addition, and of interest, phenformin is considerably more potent with an estimated IC50 of 15 μM. As already stated, phenformin was withdrawn from clinical use because of a high incidence of lactic acidosis and cardiovascular side effects.
Metformin, Sirtuin 1 and SIRT1
Sirtuin 1 is an NAD-dependent deacetylase encoded by the SIRT1 gene and is the mammalian homolog of sir2 with the latter linked to calorie- (glucose) restriction and longevity in yeast (Saccharomyces cerevisiae) and the nematode, Caenorhabditis elegans [80,81]. A potential mechanism for reducing the impact of ageing is an improvement of oxidative stress via the modulation of post-translational modification of the signalling pathways regulated by SIRT1. Sirtuins are important in the overall regulation of metabolism and their activity is associated with reducing the incidence of diabetes and other diseases that can be linked to metabolic dysregulation [82]. Deacetylation of target proteins may either enhance or inhibit activity. A very high in vivo dose of metformin, 1 g/kg, for 30 weeks, without any evidence of toxicity, extended the lifespan of middle-aged mice by approximately 6% [83]. The serum concentration of metformin in this study approached 500 μM, which is approximately 50 times greater than levels in humans, and the liver content was reported as 3.7 nM/mg protein. Of particular interest was the finding that whereas the treatment of mouse embryonic fibroblasts with metformin lowered complex 1 activity, mice treated with metformin showed an increase in complex 1 activity similar to that seen with calorie restriction. In addition, the treatment regimen also lowered indices of chronic inflammation, namely NF-κB and JNK. Inhibition by metformin of cytokine-induced activation NF-κB-mediated pathways has also been reported in endothelial cells and stem cells in breast cancer cell lines [84,85]. Inhibition of NF-κB-mediated pathways would potentially result in an inhibition of angiogenesis. Interestingly, metformin has been shown to have both pro- and anti-angiogenic activity and its pro-angiogenic actions can be linked to the protection of endothelial function and AMPK-dependent endothelial nitric oxide synthase (eNOS) regulation. Thus, in a rat heart failure model followed for 12 months, treatment with metformin at 100 mg/kg/day not only prevented the development of severe heart failure but also enhanced myocardial eNOS expression and the expression of VEGF and phosphorylated VEGF [86].
Caton et al. [87 ]reported that hepatic cells from db/db diabetic mice that were administered 250 mg/kg metformin for 7 days showed increased SIRT1 protein and activity and restored hepatic GCN5 levels, an acetyltransferase that inhibits gluconeogenesis. On the basis of these data, they concluded that metformin, via AMPK-dependent and AMPK-independent effects on SIRT1 and GCN5, inhibits hepatic gluconeogenesis. Furthermore, metformin treatment of db/db mice lowered hepatic ATP levels and enhanced the activity of AMPK; however, based on studies with hepatic hepG2 cells, metformin-mediated increases in SIRT1 protein were independent of AMPK [88]. Furthermore, adenovirus-mediated overexpression of hepatic SIRT1 in diet-induced, low-density lipoprotein receptor-deficient, insulin-resistant and insulin-resistant, genetically obese ob/ob mice corrects IR [89]. This particular study reported that overexpression of hepatic SIRT1 also attenuates the effects of a high-fat diet and obesity on the development of endoplasmic reticulum (ER) stress and signalling via the mammalian target of rapamycin (mTOR) [89]. This pathway will be discussed later in this review, but the putative pathways that mediate the effects of metformin are summarised in figure 1.
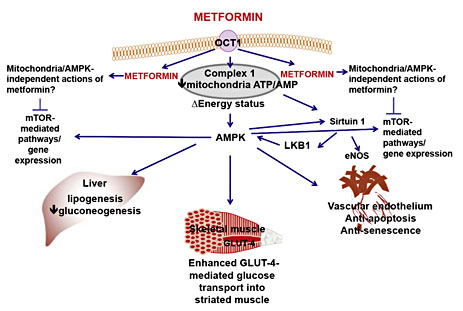
Putative cellular sites of action of metformin in the liver, striated muscle (skeletal) and vascular tissue. Metformin is a strong base with a pKa of 12.4. The expression of organic cation transporters like OCT1 is necessary for metformin absorption following oral administration as well entry into cells. Metformin can, at appropriate concentrations, inhibit mitochondria complex 1, reduce energy status and thereby lower the ATP/AMP ratio which, in turn, activates AMPK. AMPK, either directly or indirectly, inhibits liver gluconeogenesis and lipogenesis, facilitates glucose uptake into striated muscle (cardiac and skeletal) and adipose tissue (not shown). Metformin also protects the endothelium from hyperglycaemia-induced dysfunction, senescence and apoptosis. Metformin may mediate its effects indirectly on AMPK via the activation of LKB1, a serine-threonine kinase upstream of AMPK, and/or via the activation of the deacetylase Sirtuin 1, the protein product of the ‘longevity' gene SIRT1. Sirtuin 1-mediated deacetylation of eNOS enhances eNOS activity and protects endothelial cell function. An additional potential site of action for metformin is mTOR and its associated gene expression targets. Prototypically, mTOR is inhibited by AMPK, but, as depicted here, AMPK-independent mechanisms have also been described. Inhibition of mTOR may contribute to the anti-cancer activity of metformin; however, in vitro studies indicate that concentrations of metformin >1 mM are required to inhibit mTOR.
Putative cellular sites of action of metformin in the liver, striated muscle (skeletal) and vascular tissue. Metformin is a strong base with a pKa of 12.4. The expression of organic cation transporters like OCT1 is necessary for metformin absorption following oral administration as well entry into cells. Metformin can, at appropriate concentrations, inhibit mitochondria complex 1, reduce energy status and thereby lower the ATP/AMP ratio which, in turn, activates AMPK. AMPK, either directly or indirectly, inhibits liver gluconeogenesis and lipogenesis, facilitates glucose uptake into striated muscle (cardiac and skeletal) and adipose tissue (not shown). Metformin also protects the endothelium from hyperglycaemia-induced dysfunction, senescence and apoptosis. Metformin may mediate its effects indirectly on AMPK via the activation of LKB1, a serine-threonine kinase upstream of AMPK, and/or via the activation of the deacetylase Sirtuin 1, the protein product of the ‘longevity' gene SIRT1. Sirtuin 1-mediated deacetylation of eNOS enhances eNOS activity and protects endothelial cell function. An additional potential site of action for metformin is mTOR and its associated gene expression targets. Prototypically, mTOR is inhibited by AMPK, but, as depicted here, AMPK-independent mechanisms have also been described. Inhibition of mTOR may contribute to the anti-cancer activity of metformin; however, in vitro studies indicate that concentrations of metformin >1 mM are required to inhibit mTOR.
Metformin and the Microbiome
High doses of metformin, 20-100 mM, retard ageing in C. elegans by a mechanism that is partially AMPK-dependent and reported to result from the reduction of folate metabolism and the pathogenicity of bacteria in the microbiome. The anti-ageing effect of metformin was reduced by the presence of high glucose [90]. The absorption-limited pharmacokinetics for metformin could result in a local intestinal lumen concentration of metformin >1 mM, thus raising the possibility that a contributing mechanism to metformin's beneficial actions in T2DM may be to alter the microbiome [91]. It is also noteworthy that the gut microbiome differs in humans with T2DM; however, whether the microbiome in patients treated with metformin is ‘corrected' has not been reported [92].
Metformin, Glycaemic Control, Protection of the Vasculature and the Role of eNOS
NO is a potent vasodilator that, by binding to the haem moiety of guanylyl cyclase, increases cyclic guanosine monophosphate (cGMP) in vascular smooth-muscle cells (VSMCs). However, a reduction in the bioavailability of NO is associated with the development of vascular disease [93]. Interestingly, given that some of the benefits of metformin may be mediated via the modulation of SIRT1, Sirtuin 1 increases NO bioavailability by eNOS deacetylation and is involved in cell proliferation and angiogenesis [94,95]. In a cell culture protocol, 50 μM of metformin attenuates hyperglycaemia-induced reduction in ROS production and increased Sirtuin 1 expression in microvascular endothelial cells, thereby reducing apoptosis and senescence [24]. These data suggest that SIRT1 may be an important target for metformin and SIRT1-mediated endothelial cell protection via enhanced eNOS activity, a reduction in apoptosis and the promotion of angiogenesis.
Diabetes and other cardiovascular risk factors can lead to uncoupling of the eNOS dimer to the monomeric form. Uncoupled eNOS results in increased oxidative stress and eventually reduced blood flow, and a pro-thrombotic state develops. Several factors can lead to uncoupling including the up-regulation of protein levels of eNOS, which, if uncoupled, promotes the generation of superoxide anions, O2-∙ as well as the up-regulation of NADPH and xanthine oxidases. All these mechanisms increase peroxynitrite, which results in eNOS dysfunction [96]. Furthermore, hyperglycaemia increases the expression of the NADPH oxidase subunit, p22phox, and uncouples eNOS in microvascular endothelial cells [88,97].
It is therefore surprising that Zou et al. [98] proposed that metformin inhibits complex 1, resulting in the mitochondrial generation of O2-∙ and the subsequent formation of peroxynitrite, the latter mediating the activation of AMPK in bovine aortic endothelial cells. The effects of metformin were blocked by adenoviral overexpression of uncoupling protein, UCP-1, which blocks electron transfer and the generation of O2-∙. The metformin-mediated activation of AMPK, but not activation due to the AMPK agonist, AICAR, was absent in the aorta and heart from eNOS-/- mice. These data are seemingly in conflict with other studies that found that metformin improves the bioavailability of NO and endothelial function; the possible explanation is that the threshold concentration of metformin required to activate AMPK is 100 μM, and a maximal effect has been reported with 1 mM. Of interest, therefore, are the data from a study of the effects of metformin in bovine aortic endothelium that demonstrate a concentration-dependent increase of 50-1,000 μM in eNOS activity and serine 1179 phosphorylation, but a decline in cGMP levels, with the highest metformin concentration suggesting pro-oxidative effects [99].
Metformin has been shown to have favourable haemodynamic and rheological effects in patients with diabetes and other cardiovascular risk factors. A study examining the effect of L-arginine infusion, the substrate for eNOS and the molecular source for the generation of NO in diabetics showed enhanced haemodynamic effects following treatment with metformin [100]. The blood flow in muscle and adipose tissue is also increased following treatment with metformin [101]. Another study reported lower systolic blood pressures in response to vasoconstrictors, such as norepinephrine and angiotensin II, in patients treated with metformin in comparison to those treated with the sulphonylurea or glibenclamide despite similar glycaemic control [102]. Significant improvement in acetylcholine-mediated vasodilation has also been documented with metformin therapy in T2DM patients [23]. Similar findings have been reported for the effects of low-dose metformin, 500 mg b.i.d., on endothelial function in patients with metabolic syndrome [103]. However, the cellular mechanism(s) whereby these favourable effects of metformin are mediated is still unclear. Thus, the beneficial effects of metformin could be entirely secondary to a reduction in IR [103]. Other mechanisms have been hypothesised that imply that metformin has a direct action on the endothelium and/or vascular smooth muscle; for instance, antioxidant effects, an improved contribution of NO to EDV, effects on calcium homeostasis, AMPK-mediated effects, the inhibition of mitochondrial complex 1 or the inhibition of apoptosis [24,104,105,106,107]. Thus, the results from a study by Detaille et al. [107] with human dermal cells, human umbilical vein cells and bovine aortic cells complement the data already discussed from the laboratories of El-Mir et al. [65] and Owen et al. [67]. Detaille et al. [107] demonstrate that high concentrations of metformin inhibit complex 1 in digitonin-permeabilised endothelial cells and prevent high-glucose-induced endothelial cell death via the inhibition of the opening of the mitochondrial permeability transition pore. High concentrations of metformin, e.g. >500 μM,have also been shown to inhibit the expression of mRNA and protein for thioredoxin-interacting protein, Txnip [108]. In this particular study, HeLa cells were exposed to 2 mM of metformin, and the data indicate a very rapid effect of metformin on mRNA levels of Txnip with a significant reduction noted at 1 h. Txnip is an inhibitor of the redox regulator, thioredoxin, and is associated with elevated oxidative stress. The expression of Txnip is enhanced by high glucose and overexpression can result in beta-cell apoptosis [109]. As already discussed, certain concentrations of metformin have been reported to be toxic to cell function [33].
Acetylcholine-mediated EDV is improved in third-order small mesenteric arteries from fructose-induced, insulin-resistant rats following acute treatment in vitro with 100 μM of metformin or chronic treatment in vivo for 2 weeks with 300 mg/kg of metformin [105]. These data suggest that metformin does have a direct action on endothelial cells and indicate that the beneficial effect(s) of metformin on endothelial function is (are) NO-dependent. Of interest is that, in the same study, it was also reported that high concentrations of metformin, e.g. >1 mM, produced an endothelium-independent contractile action in isolated mesenteric arteries from the rats, suggesting a direct action of metformin on calcium homeostasis. This interpretation is supportive of data from another study on the effects of metformin on rat-tail VSMCs [110]. Similarly, in high-fat-fed Goto-Kakizaki rats, an animal model of non-obese T2DM, treatment with 60 mg/kg/day of oral metformin significantly lowered the levels of IR, AGEs, oxidative stress and the plasma inflammatory marker CCL2, and enhanced the bioavailability of NO and improved EDV in the thoracic aorta [111]. Verma et al. [112 ]reported that a 6-week treatment with high-dose metformin of 500 mg/kg/day prevented the development of fructose-dependent hypertension, IR and hyperinsulinaemia, and also restored EDV and insulin-mediated vasodilatation; these data suggest that the benefits of metformin are secondary to the improvement of vascular sensitivity to insulin. Using an isolated perfused rabbit kidney preparation, Gomes et al. [113] reported that 20-100 μM of metformin prevented the reduction in acetylcholine-mediated EDV which resulted from an acute 3-hour infusion that raised the glucose concentration from 5.5 to 15 mM. Isoda et al. [114 ]reported that a 1-hour exposure of human saphenous vein endothelial cells to 20 μM of metformin prevented high-glucose-induced (e.g. 30 mM) phosphorylation of Akt and PKC. Metformin-mediated phosphorylation of AMPK was absent with 2, 20 and 200 μM, however, and was only observed when the metformin concentration was raised to 2 mM. In contrast, 1 mM of metformin was required to inhibit IL-1β-induced phosphorylation of Akt and, ultimately, the translocation of the pro-inflammatory transcription factor, NF-κB, in isolated smooth-muscle cells from human saphenous veins. A similar discrepancy in sensitivity was noted for the ability of metformin to inhibit IL-1β-induced IL-6 and IL-8 cytokine production in macrophages, endothelial cells and VSMCs. The VSMCs were far less sensitive to metformin than the endothelial cells and macrophages.
The vascular protective effects of metformin are not limited to the endothelium. Studies suggest that metformin has anti-hypertensive effects via a direct action on VSMCs. A study examining the anti-hypertensive effects of therapeutically appropriate concentrations of metformin (15 μM) showed that treatment of aortic smooth-muscle cells from the spontaneously hypertensive rat led to a decrease in thrombin and vasopressin-induced elevations in intracellular calcium [106].
Metformin, Vascular Protection and Insulin Resistance
The role of IR in the development of endothelial dysfunction is controversial. Some studies have not been able to find a relationship between IR and endothelial dysfunction in patients with cardiovascular risk factors such as obesity [115]. However, several cross-sectional studies have demonstrated a correlation with endothelial dysfunction in obese subjects [116], T2DM subjects [117] and those with essential hypertension [118]. The thiazolidinedione and peroxisome proliferator-activated receptor γ ligand troglitazone, also an insulin-sensitiser, improves IR in obese subjects, but has no effect on vascular responses [119]. In contrast, treatment with metformin improves both IR and endothelium-dependent vascular responses in diabetic patients [23]. Collectively, these data strongly suggest that metformin protects the vasculature through mechanisms that are independent of IR. However, a study with another peroxisome proliferator-activated receptor γ ligand, pioglitazone, concluded that this thiazolidinedione did improve both insulin sensitivity and EDV as well as elevating blood levels of NO in patients with impaired glucose regulation [120].
Metformin and the Regulation of Glucose Metabolism in Endothelial Cells
Glucose metabolism in endothelial cells is predominantly glycolytic; however, with hyperglycaemia, glucose can be shunted into other pathways such as the polyol and hexosamine pathways as well as being directed to the non-enzymatic generation of AGEs and PKC [28]. Except for the potential direct effects of metformin on AGEs, there does not seem to be any convincing evidence that metformin interferes with such pathways.
Metformin and AGEs
Metformin has been shown to chemically interact with the AGE precursors glyoxal and methylglyoxal to form guanidine-dicarbonyl adducts and thus reduce the production of AGEs [121]. Studies were performed with equimolar concentrations of glyoxal, methylglyoxal and metformin, and suggest that metformin, in addition to other actions, may also serve as a scavenger of dicarbonyl AGE precursors. It is, however, not clear to what extent this mechanism contributes to the vaso-protective properties of metformin.
PKC Activation
Via the polyol pathway, an accumulation of triose phosphate activates PKC, an important family of at least 12 isoforms of proteins that modulate several intracellular signalling cascades by phosphorylating serine/threonine amino acids on several functional proteins. Most PKC isoforms are activated by diacyl-glycerol, the levels of which increase in a hyperglycaemic milieu [28]. Activation of PKC can affect vascular blood flow by several mechanisms; for instance PKC-β has been shown to play a role in renal and retinal blood flow problems, possibly by depressing NO production and increasing endothelin-1 activity [122]. Gallo et al. [123] demonstrated that the in vitro treatment of human umbilical vein endothelial cells with therapeutically appropriate concentrations of metformin, i.e. 20-40 μM, attenuated the hyperglycaemia-induced translocation of PKC-β2 from the cytosol to the membrane and was associated with an increase in the expression of the hydrogen peroxide-scavenging enzyme catalase as well as a reduction in NADPH activity.
In conclusion, hyperglycaemia, either directly or indirectly via AGE- or PKC-mediated pathways, enhances oxidative stress and triggers endothelial dysfunction, ER stress and the development of vascular disease, and these processes are directly and/or indirectly modulated by metformin.
A summary and brief critique of the major putative cellular targets for metformin are given in table 1.

Mammalian Target of Rapamycin
PKC is also downstream of mTOR and regulates gene expression of the enzymes involved in mitochondrial oxidative metabolism [124]. mTOR plays a part in the regulation of multiple cellular pathways involved in cell growth, proliferation, mitochondrial oxidative function and cell survival. It is of interest that rapamycin, which inhibits mTOR, has been shown to extend life expectancy in mice [125]. As already discussed, metformin also extends life expectancy in mice [83] and metformin either directly, or indirectly via AMPK, targets mTOR, albeit at mM concentrations. mTOR is a serine/threonine protein kinase that is activated in high-energy states and, conversely, is inhibited under conditions of energy depletion; the latter is mediated, in part, via the AMPK-mediated inhibition of Raptor, a protein that through its interaction with mTOR has a positive role in nutrient-stimulated signalling [126,127]. AMPK-independent negative regulation of mTOR via REDDI has also been reported. Thus, activation of AMPK does not seem to be an absolute requirement for mediating the cellular actions of metformin [128]. Inhibition of mTOR also promotes autophagy (autophagocytosis or ‘self-eating') that enables tissues to maintain basal activity despite nutrient deprivation and occurs after ER stress. ER stress can be initiated by hypoxia, glucose deprivation (e.g. following a myocardial infarction), severe hypoglycaemia and hyperglycaemia, and these are all conditions that can prevail in patients with diabetes [129].
The metformin-mediated inhibition of the mTOR pathway and promotion of autophagy and apoptosis may also explain the putative anti-cancer efficacy of this drug [130]. As previously discussed, metformin can have both positive and negative effects on angiogenesis. An inhibitory effect would reduce tumour growth [6,84,85,131]. In in vitro studies, metformin treatment of women with polycystic ovary syndrome has been reported to increase the production of anti-angiogenic thrombospondin-1, indicating effects that are mediated via the inhibition of NF-κB and Erk1/2/Erk5 signalling [132]. Inhibition of mTOR by rapamycin has been shown to have a protective action against the development of atherosclerotic plaques; however, the use of rapamycin has also been associated with hyperglycaemia whereas metformin normalises glycaemia [133,134]. Thus, metformin-mediated inhibition of mTOR may be a contributory factor to the overall cardiovascular protective actions of metformin, but it is not clear whether this necessarily involves the activation of AMPK. The role of the mTOR signalling pathway in mediating any of the therapeutic actions of metformin is questionable given that, in order to modulate mTOR signalling in vitro, metformin concentrations in excess of 1 mM are required. In vitro studies have demonstrated metformin-induced cell death with pancreatic beta cells [33,135] as well as with an acute lymphoblastic leukaemia cell line, but the concentrations of metformin required for this are >1 mM. Pancreatic beta-cell dysfunction is not associated with the long-term therapeutic use of metformin and thus the interpretation of in vitro studies that have utilised such high concentrations of metformin is difficult, particularly when one attempts to extrapolate to the use of metformin in a therapeutic dose range.
Conclusion
Metformin is a valuable drug for the treatment of T2DM, with minimal significant side effects. It provides benefits beyond its action as an oral hypoglycaemic agent and insulin sensitiser. Data from both clinical and bench studies indicate that metformin has a direct action on the endothelium and thus provides protection against the development of hyperglycaemia-induced vascular disease. The cellular mechanisms involved in the vasoprotective effects of metformin and its actions in the liver and skeletal muscle still have to be further elucidated, but seem to involve a reduction in oxidative stress that may or may not be secondary to an action on mitochondrial complex 1. They possibly involve the activation of AMPK and/or other pathways, such as those mediated by the deacetylase Sirtuin 1 and the regulation of eNOS. Although there is a vast dataset on metformin-mediated activation of AMPK being a contributor to the insulin-sensitising and hepatic gluconeogenesis inhibitory actions of metformin, some data are contradictory, suggesting that AMPK-independent actions are also important. The wide range of concentrations used in in vitro studies has contributed to the confusion in the literature regarding the role of AMPK inhibition of complex 1 and the mTOR pathway in mediating the therapeutic effects of metformin. Many drugs show concentration-dependent effects on multiple targets, but such targets do not necessarily represent the therapeutic target(s) for the drug. Although no evidence of significant levels of metabolites of metformin has been detected, it remains a possibility that metformin is a pro-drug and, as suggested by Hawley et al. [63], intracellular conversion to a low level of a minor metabolite occurs and the time-dependent accumulation of this putative metabolite mediates the effects of metformin. Thus, despite almost 60 years of research, we still do not fully understand the cellular mechanisms that underlie the important therapeutic effects of metformin. However, we can conclude that the use of metformin in T2DM does have both direct and indirect benefits on vascular function.
To paraphrase a conversation between Piglet and Pooh in Winnie the Pooh by A.A. Milne [136] that concerns Pooh and his hunting for something: ‘We are indeed tracking the cellular mechanisms of action of metformin, but where this hunt will lead us is still unclear.' The in vivo and in vitro studies of metformin have taken us down many paths that reflect the dose/concentration, time and tissue effects of this interesting compound. Which of these sites and mechanisms of action, if any, truly explain the therapeutic benefits of metformin still has to be determined. Indeed, we still need to hunt down the cellular mechanism(s) by means of which metformin protects the vasculature against glucose toxicity. Such knowledge will enable us to design even better therapeutic interventions for the prevention of the high cardiovascular morbidity associated with diabetes. In this regard, the new field of metabolomics is potentially of value and the analysis of metformin-induced changes in the molecular milieu of patients treated with the drug may suggest novel targets for the action of this widely used hypoglycaemic and vascular protective agent [137].

