

Abstract
Recent progress in psychiatric research has accumulated many mouse models relevant to developmental neuropsychiatric disorders using numerous genetic and environmental manipulations. Since the prefrontal cortex (PFC) is essential for cognitive functions whose impairments are central symptoms associated with the disorders in humans, it has become crucial to clarify altered developmental processes of PFC circuits in these mice. To that end, we aimed to understand a sequence of molecular events during normal mouse PFC development. Expression profiles for representative genes covering diverse biological processes showed that while there were little changes in genes for neuroreceptors and synaptic molecules during the postnatal period, there were dramatic increases in the expression of myelin-related genes and the parvalbumin gene, peaking at postnatal day (P)21 and P35, respectively. The timing of the peaks is different from that observed in the striatum. Furthermore, the evaluation of the circuitry maturation by measuring extracellular glutamate in the PFC revealed that sensitivity to an NMDA antagonist did not become an adult-like pattern till P56, suggesting that some of the maturation processes continue till P56. The trajectory of molecular events in PFC maturation described here should help us to characterize how the processes are affected in disease model mice, an important first step for translational research.
Introduction
The prefrontal cortex (PFC) is a pivotal brain region for executive cognitive functions, including working memory, planning, reasoning, and decision-making [1]. Acquisition of these higher brain functions depends on the development and maturation of the PFC, which take decades to complete in humans [2,3,4]. PFC development and maturation involve diverse biological processes, namely synapse formation, local circuitry formation, connectivity among several brain regions through long projections (network formation), the formation of neuromodulation networks, activity-dependent synaptic modulation and plasticity, and myelination. These processes should proceed in a coordinated and sequential manner, each process affecting others [5].
Impairments in cognitive functions are central symptoms associated with developmental neuropsychiatric disorders such as schizophrenia [6]. As a potential etiology of schizophrenia, developmental hypothesis has been proposed, in which combinations of genetic and environmental factors cause alterations in developmental processes that serve as the biological basis for pathogenesis of the disorder [7,8]. In schizophrenia, PFC development is presumed to be disrupted, resulting in cognitive impairments even before the onset of the disorder (the so-called prodromal period) [9]. Brains with schizophrenia also show changes associated with many systems including dopamine, glutamate, GABA, and myelin [10].
Recent human genetics studies have provided us with a list of candidate genes involved in disorders like autism and schizophrenia [11,12,13]. Subsequently, a number of mouse models with modification of those genes have been developed, many of which display behavioral phenotypes relevant to human psychiatric disorders [14,15,16]. To understand the biological processes affected in these animals that can be translated into cognitive impairments in the disorders in humans [17], it is crucial to understand a developmental trajectory of the PFC in mice [5]. In the present study, we characterized a sequence of molecular events in the developing mouse PFC using gene expression profiles as molecular signatures for developmental processes.
Materials and Methods
Animals
All animals used in this study were treated in accordance with the guidelines by the Animal Care and Use Committee of Kyoto University and Johns Hopkins University. Wild-type C57BL/6JJcl mice were purchased from CLEA Japan (Tokyo, Japan) for qRT-PCR and from Charles River Laboratories (Wilmington, Mass., USA) for in vivo microdialysis. PV/myrGFP-LDLRct mice in the C57BL/6JJcl background that express myristoylated green fluorescence protein (GFP) fused with the low-density lipoprotein receptor (LDLR) cytoplasmic region under the parvalbumin (PV) gene promoter were previously described [18].
RNA Preparation and qRT-PCR Analysis
Brain regions of interest were identified using the atlas of Paxinos and Franklin [19] and collected from 1-mm-thick brain slices using a tissue puncher with a diameter of 1.2 mm (online suppl. fig. 1, www.karger.com/doi/10.1159/000430095). Total RNAs from the medial PFC and striatum were extracted using the RNeasy Mini kit (QIAGEN, Hilden, Germany), and cDNAs were synthesized using the ReverTra Ace qPCR RT Master Mix (TOYOBO, Osaka, Japan). qRT-PCR was performed using the 7900HT Fast Real-Time PCR System or ViiA™7 Real-Time PCR System (both Applied Biosystems, Waltham, Mass., USA) with the THUNDERBIRD Probe qPCR Mix (TOYOBO) and Universal ProbeLibrary (Roche, Indianapolis, Ind., USA) [20]. Primers were designed using the Universal ProbeLibrary Assay Design Center (Roche; table 1). All samples were run in triplicate. Relative gene expression levels to the level at postnatal day (P)7 were calculated using the ∆∆Ct method normalized with Gapdh as an endogenous control gene.

Histology
Mice were anesthetized with a mixture of ketamine and xylazine and transcardially perfused with phosphate-buffered saline (PBS) followed by 4% paraformaldehyde in PBS. The isolated brains were postfixed overnight, and 50-μm-thick coronal sections were prepared using a vibratome VT1200S (Leica Biosystems, Buffalo Grove, Ill., USA). The sections were washed with PBS and mounted with VECTASHIELD mounting medium with DAPI (Vector Laboratories, Burlingame, Calif., USA). Fluorescent images were acquired using an all-in-one fluorescence microscope BZ-9000 (KEYENCE, Osaka, Japan). We counted all GFP-positive cells in both layer 2/3 and layer 5/6 of area 24 in all sections ranging from a brain section containing the rostral end of the corpus callosum connecting two hemispheres to a brain section containing the posterior crus of the anterior commissure connecting two hemispheres.
Microdialysis
Microdialysis was carried out as previously described [21], with minor modifications. A guide cannula (AG-6, Eicom, Kyoto, Japan) was implanted into the frontal cortex [15° angle, anteroposterior from the bregma, +1.7 mm (P56 and P140), +1.65 mm (P42), +1.5 mm (P28); mediolateral from the bregma, -1.0 mm (P56 and P140), -0.9 mm (p42), -0.6 mm (P28); dorsoventral from the dura, -2.0 mm (P56 and P140), -1.8 mm (P42), -1.65 mm (P28)] according to the atlas of Paxinos and Franklin [19]. Ringer's solution (147 mM NaCl, 4 mM KCl, and 2.3 mM CaCl2) was perfused at a flow rate of 1.0 µl/min. The samples were collected every 10 min from awake, freely moving mice and analyzed by an HPLC system (Eicom). Six samples were taken to establish the baseline measurement of extracellular glutamate. The levels of extracellular glutamate were analyzed after the intraperitoneal injection of MK-801 (1 mg/kg).
Results
To gain insights into the biological events during PFC development, we performed gene expression analysis by qRT-PCR, targeting representative genes for multiple neural components, namely, neurotransmitter receptors, synaptic components, GABAergic interneurons, and oligodendrocyte/myelin (table 1). We chose five postnatal time points: P7, P14 (two time points before weaning), P21 (weaning), P35 (adolescence), and P63 (adulthood).
Glutamate Receptors and Synaptic Components in the Developing PFC
Neurotransmitter receptors are central players for neurotransmission and critical for synaptic functions. We focused on two types of glutamate receptors, NMDAR and AMPAR, two major receptor types for excitatory synaptic transmissions. None of the AMPARs analyzed showed notable changes in their expressions during the course of development and maturation (fig. 1a). The NMDARs are heterotetrameric complexes comprised of two NR1 and two NR2 subunits; and among the NR2s, NR2A and NR2B are the predominant subunits expressed in the cortex. Grin1 showed a 4-fold reduction in its expression at P14 compared to P7 and stayed at this level, whereas Grin2a showed a 4-fold increase in its expression at P14 and stayed at that level. Grin2b showed a gradual decrease in its expression, though not so drastically throughout development (fig. 1b). It has been observed that changes in the composition of NMDAR subunits, namely switching from NR2B to NR2A, take place at the early postnatal stage [22]. The gene expression patterns we observed may correspond to this switching event. Next, we characterized gene expression patterns for a neuromodulatory system, namely dopamine receptors (DRDs), which are targets of most antipsychotics effective for positive symptoms of schizophrenia [23]. Among the 5 DRDs, we could not detect sufficient amplification signals in the PFC for Drd3 which has been shown to be predominantly expressed in limbic regions of the brain [24]. Most of the DRDs analyzed showed a slight reduction in their expressions except Drd4 which showed a slight increase in its expression during development (fig. 1c).

Gene expression profiles in the developing PFC. a AMPARs, b NMDARs, c DRDs, d PSD molecules, e CAMs, f interneuron-related genes, and g oligodendrocyte/myelin-related genes (see table 1). The endogenous control gene, Gapdh, was used for normalization. Data are the means of three independent samples for each point.
Gene expression profiles in the developing PFC. a AMPARs, b NMDARs, c DRDs, d PSD molecules, e CAMs, f interneuron-related genes, and g oligodendrocyte/myelin-related genes (see table 1). The endogenous control gene, Gapdh, was used for normalization. Data are the means of three independent samples for each point.
We characterized several synaptic components as markers for synaptic development and maturation. Scaffolding proteins at the postsynaptic density (PSD) are crucial structural components, important for anchoring synaptic proteins like channels, receptors, and synaptic cell adhesion molecules (CAMs) [25]. We chose two representatives, Psd95 and Shank3, because recent findings have suggested that there might be differential synaptic localizations of these molecules, at least in humans [26]. Fmr1 and Cyfip1 are important translational modulators at synapses whose alterations are associated with developmental neuropsychiatric disorders [27]. All these 4 genes did not show any notable differences at all 5 time points (fig. 1d). CAMs are key players for supporting synaptic connections [28] and are also involved in neuron-glial interactions, e.g., myelination [29]. Most of the CAMs analyzed did not show drastic changes in the expressions during the development of the PFC except Cntnap1 which plays a role in the formation of myelinated nerve structures [30]. Cntnap1 increased its expression at P14 by 4 fold compared to P7 and gradually increased its expression thereafter (fig. 1e).
GABAergic Inhibitory Interneurons in the Developing PFC
GABAergic inhibitory interneurons are essential components for local circuitry. Proper excitatory/inhibitory balance, determined by the activities of excitatory and inhibitory neurons, is crucial for regulating an activity of the local circuitry that serves as a module in the neuronal networks, necessary for higher brain functions [e.g., [31,32]]. Although all of these interneuron populations share a common character, i.e., use GABA as their neurotransmitter, they are comprised of diverse groups of neurons. Recent studies have classified GABAergic interneurons based on morphology, molecular profiling, and electrical properties into 3 major subclasses such as PV-positive fast-spiking neurons, somatostatin (Sst)-positive burst-spiking neurons, and vasoactive intestinal peptide (Vip)-positive regular-spiking neurons [33]. When we characterized the expression of the glutamate decarboxylase 1 (Gad1) gene, a marker gene for all GABAergic interneurons, we did not detect dynamic changes during development. However, when we analyzed the cell type-specific profiles, we found quite different pictures. The expression of the PV gene was drastically increased between P7 and P21 (about 256 fold) and reached its peak level at P35. The Vip gene showed a 4-fold increase between P7 and P21, and peaked at P35. The Sst gene did not show drastic changes (fig. 1f). These data suggest that the appearance of the PV-positive neurons in the PFC drastically increases between P7 and P21. To confirm this, we analyzed transgenic mice expressing myristoylated GFP under the promoter of the PV gene. The PV-positive interneurons comprised of basket cells and chandelier cells are localized in the upper (layer 2/3) and lower (layer 5/6) layers in the mouse PFC. Consistent with the gene expression data above, the total number of GFP-positive cells increased between P21 and P35 in both layer 2/3 and 5/6 in the PFC (fig. 2).

Appearance of PV-positive cells in the developing PFC. The total number of GFP-positive (GFP+) cells in layer 2/3 (a) and layer 5/6 (b) of the anterior cingulate cortex (area 24) of PV/myrGFP-LDLRct mice that express GFP under the PV gene promoter were counted as described in Materials and Methods. Data are the means ± SEM of two independent samples for each point.
Appearance of PV-positive cells in the developing PFC. The total number of GFP-positive (GFP+) cells in layer 2/3 (a) and layer 5/6 (b) of the anterior cingulate cortex (area 24) of PV/myrGFP-LDLRct mice that express GFP under the PV gene promoter were counted as described in Materials and Methods. Data are the means ± SEM of two independent samples for each point.
Myelin and Oligodendrocytes in the Developing PFC
A local circuitry on its own serves as a module that needs to be connected with other modules spread over the brain in order to function in the networks. This is achieved by long projections connecting among these modules. This configuration is the basis for neural networks that are crucial for behavioral tasks [34]. Long projections support a fast neural transmission along the pathways to ensure proper information processing through networks, which is achieved by their myelination by oligodendrocytes [35]. Transmitted signals through the myelinated long projections affect the nature of inputs to the local circuitry, modulating the maturation of the local circuitry's activity; and, therefore, myelination/oligodendrocyte development in the PFC must be correlated with the maturation processes of the PFC. We characterized expression profiles of several oligodendrocyte/myelin-related genes in the developing PFC. Two transcription factors, Olig2 and Sox10, controlling the differentiation of oligodendrocytes [36,37] did not show significant changes. However, myelin components, such as Plp1 and Mag, showed drastic increases (64 fold) between P7 and P21 and reached their peak level at P21. Other components important for the formation of the myelinated nerve structure, such as Cldn11 and Cnp1, also showed increases (4-8 fold) with a similar time course (fig. 1g).
MK-801 Sensitivity of the Developing PFC
The results above suggest that PFC maturation may continue at least till P35, when the PV gene expression reaches the maximum level. The systemic administration of an NMDAR antagonist, MK-801, increases the glutamate efflux in the PFC [38], and the sensitivity to NMDAR antagonists is thought to reflect physiological properties of the neural circuitry [e.g., [39]]. Therefore, to clarify the timing of the functional maturation of the PFC, we investigated the responsiveness to the administration of MK-801 by measuring the extracellular glutamate level in mouse PFC using in vivo microdialysis. We analyzed four different time points: P28, P42, P56 (three time points during adolescence), and P140 (adulthood). The level of extracellular glutamate was lower at P28 compared to the adult level, but reached a similar level to that in adult by P42 (data not shown). However, the response of glutamate release to the administration of MK-801 was more exaggerated at P42 than the response in adult. At P56, the response became similar to the one at P140 (fig. 3). These results suggest that functional circuitry maturation in the PFC involving a NMDAR may be achieved by P56, far beyond P42.
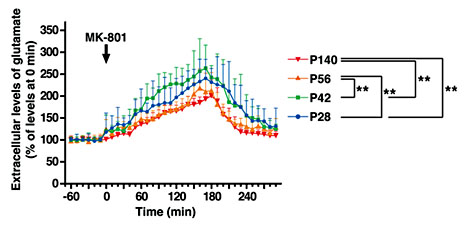
Measurement of extracellular glutamate in the frontal cortex. The changes in the extracellular glutamate level after the administration of MK-801 were monitored by in vivo microdialysis. Data are means ± SEM (n = 10-14 for each group). Statistical significance was determined using one-way ANOVA and subsequent Fisher's PLSD for multiple comparisons. ** p < 0.01.
Measurement of extracellular glutamate in the frontal cortex. The changes in the extracellular glutamate level after the administration of MK-801 were monitored by in vivo microdialysis. Data are means ± SEM (n = 10-14 for each group). Statistical significance was determined using one-way ANOVA and subsequent Fisher's PLSD for multiple comparisons. ** p < 0.01.
Gene Expression Patterns during the Postnatal Development of the Striatum
Since the striatum is another place that is presumed to be affected in developmental neuropsychiatric disorders like schizophrenia [40], it was of interest to compare developing patterns in the striatum to the ones in the PFC. We performed the same gene expression analyses using mouse striatum during the same time periods as above. The patterns of AMPARs, NMDARs, and synaptic components were more or less the same as the ones observed in the PFC (fig. 4a, b, and d). The DRDs showed similar patterns except Drd4 which showed a more pronounced reduction at P21 (fig. 4c). The CAMs also showed similar trends of the expression changes (though there were some quantitative differences) during development (fig. 4e). However, the expression of the interneuron genes showed quite different patterns. There were 16-fold increases both in the PV and Vip gene expressions between P7 and P14, but the expressions decreased thereafter (fig. 4f). Furthermore, the oligodendrocyte/myelin-related genes showed quite distinct patterns from those in the PFC. Whereas Olig2 and Sox10 showed similar patterns to the ones in the PFC, genes such as Plp1, Mag, Cldn11, and Cnp1 increased in their expressions and reached their maximum levels by P14 (fig. 4g).

Gene expression profiles in the developing striatum. a AMPARs, b NMDARs, c DRDs, d PSD molecules, e CAMs, f interneuron-related genes, and g oligodendrocyte/myelin-related genes (see table 1). The endogenous control gene, Gapdh, was used for normalization. Data are the means of three independent samples for each point.
Gene expression profiles in the developing striatum. a AMPARs, b NMDARs, c DRDs, d PSD molecules, e CAMs, f interneuron-related genes, and g oligodendrocyte/myelin-related genes (see table 1). The endogenous control gene, Gapdh, was used for normalization. Data are the means of three independent samples for each point.
These results implicate that expression changes during development show regional differences, and that the PFC seems to have a slower time course of maturation than the striatum, based on the expression of interneuron and myelin-related genes. It has been shown that neuronal maturation as well as myelination in the human PFC is slower compared to any other brain region [41,42]. Our data support that this may be applied to the rodent PFC as well.
Discussion
Developmental neuropsychiatric disorders such as schizophrenia show impairments in cognitive functions that are mediated by the PFC (e.g., working memory [43]). The developmental processes of the PFC may be affected by a combination of genetic and environmental factors, serving as biological bases for pathogenesis of the disorder [8,44]. The establishment of functional neural circuits requires temporally coordinated, multiple biological processes, including neurogenesis, the migration of newborn neurons, neurite outgrowth, gliogenesis, synaptogenesis, myelination, and synapse maturation/elimination [45]. In the present study, we focused on the processes taking place postnatally, namely synaptogenesis, activity-dependent synaptic modulation, the development of neuromodulation (dopamine), and myelination during the development and maturation of the PFC, focusing on processes relevant to pathophysiology of the disorders.
Gene Expression Profiles in the Developing PFC
Neuronal activity in the local circuitry depends on glutamatergic neurons. It has been shown that during maturation there is a subunit switch in NR2 components, namely NR2A and NR2B [22]. We detected changes in the expression of genes for these subunits at early postnatal ages that may reflect this subunit switch. However, at P14, the glutamate receptors already showed similar expression patterns to the ones in the mature brain.
Cognitive functions involving the local circuitry require maintaining appropriate excitatory/inhibitory balance [e.g., [46]], in which GABAergic interneurons play crucial roles. In the present study, individual GABAergic interneuron populations showed distinct patterns of the gene expression profile. Especially, the PV gene showed a unique pattern, drastically increasing its expression with a peak at P35. The maturation of PV neurons depends on appropriate inputs from pyramidal neurons [47], a process dependent on NMDARs on the PV neurons [48]. These data imply that the maturation of the PV neurons is correlated with local circuitry maturation. Local circuitry maturation in turn should be correlated with functional measurements of the PFC, such as gamma oscillation [49]. Altered gamma oscillation is reported in schizophrenia [32], possibly underscoring the importance of maturation of the PV neurons in schizophrenia pathogenesis.
When we characterized the expression patterns of the oligodendrocyte/myelin-related genes, we found that the Mag and Plp1 genes, whose products are directly involved in myelin sheath formation, notably increased in their expressions during the development of the PFC. It has been shown that the degree of myelination depends on the neuronal activity of the axons that are wrapped by oligodendrocytes [50]. These data support the idea that maturation of neuronal activity in the PFC circuits induces maturation of myelination of the PFC neurons. In addition, the degree of myelination should also affect maturation of the local circuitry's activity, having bidirectional interactions. A recent report demonstrating that complicated motor skill learning requires active central myelination in the corpus callosum in adults [51] has underscored the importance of myelination in maturation of brain functions.
When we characterized the extracellular glutamate level in the PFC, we observed a similar level already at P42 to adulthood. However, the response of glutamate release in the frontal cortex to the systemic administration of MK-801 was still exaggerated at P28 and P42, and became similar to the adult level at P56. This suggests that although gene expression patterns of glutamate and inhibitory neurons as well as myelination become similar to the adult ones at P35, the circuitry is still functionally immature. At this moment, we do not know what correlates with this time period that is vulnerable to MK-801, gene expression wise, but possible candidates of biological processes taking place during this late maturation period would be ongoing synaptic pruning, the functional maturation of inhibitory neurons, and/or the maturation of neuromodulation such as the dopamine system.
Behaviorally, mice exhibit developmental milestones reflecting the acquisition/disappearance of different abilities/tendencies at certain ages that have been described in many behavioral tasks such as novel object recognition tasks [52]. At P21, mice demonstrate object exploration comparable to adults, but they are unable to detect either object rearrangement or object novelty. At P28, mice show higher locomotion activity and object exploration and are able to detect the spatial rearrangement, though they do not display preference for the displaced objects (which adult mice would do). They also clearly react to object novelty. At P49 (which is equivalent to adolescence), mice show the highest levels of object exploration and are able to detect spatial rearrangement, but they still do not display any preference for the displaced objects. They also react to object novelty. Although spatial learning is mediated by the hippocampus, some of these features may also be associated with PFC development. Moreover, it has been shown that at P42-P56, mice can perform well in Y-maze and object-based attention tests [53], supporting the idea that abilities of working memory and attention, features mediated by the PFC, are established around this time period.
As for social behavior, another brain function involving the PFC, adults are mainly concerned with dominance/submission and reproductive matters, spending most of their social time in patrolling, fighting, copulating, and raising litters. However, developing mice (namely at P21-P45) engage in frequent and prolonged bouts of amicable and playful behavior [54]. This complex and subtle domain of social activity seems to have a prominent role in the development and refinement of species-specific behavioral repertoire (from motor to cognitive and social skills) [54]. The development of some of these social behaviors may also be associated with PFC development.
When we compared gene expression profiles of the developing PFC with those of the developing striatum, the timing of the peaks was different for the interneuron and myelin genes (compare fig. 1 and 4). Furthermore, even in the cerebral cortex, the patterns seemed to be different from the ones in the motor and sensory cortices (unpubl. results). These results suggest that the PFC has unique developmental profiles of gene expression, perhaps reflecting a different timing of the maturation of biological processes.
Gene Expression and Functional Outcomes: Molecular Targets for Translation
The gene expression profiles can be used as molecular signatures of biological processes during PFC development. Many mouse models with modified genes that are associated with developmental neuropsychiatric disorders like autism and schizophrenia have been developed and characterized. Analyses of the PFC in those mice showed a wide range of changes including synaptic dysfunction, decreased PV interneurons, alterations in the excitatory/inhibitory balance, and myelination deficits [e.g., [20,55,56,57,58,59,60,61,62,63]]. Using the molecular signatures described here, it will become possible for us to identify pathological chains of events introduced in those animals, which may lead to behavioral alterations, namely cognitive deficits. Once we identify critical biological events that are affected in these mouse models, we could devise therapeutic approaches to correct those biological events, by which we may be able to improve behavioral deficits [e.g., [64,65,66,67,68,69]].
Interestingly, the onset of schizophrenia in humans is usually in the late teens and early twenties. It is believed that during adolescence, the brain receives impacts from environmental insults, resulting in a dysfunction in the system that manifests as psychosis episodes (the onset of the disorders). In the case of mice, adolescence corresponds to the time period from weaning to adulthood (P28-P56). The PFC shows an extended period sensitive to the detrimental effects of stress exposure [70], and our data suggest that the time window for environmental insults to the PFC remains open at least till P56 in mice. It has been shown that social isolation during P21-P35 affected PFC myelination and induced behavioral alterations, whereas social isolation during P35-P56 did not affect PFC myelination nor did it induce those behavioral alterations observed in the former isolation condition [71]. Interestingly, social isolation during P35-P56 affected the gene expression patterns in the mesocortical system, but not in the mesolimbic dopaminergic system through the hypothalamic-pituitary-adrenal (HPA) axis, i.e., stress response, and showed behavioral alterations in different domains [21]. The former time period corresponds to the period of myelin maturation (P21-P35), whereas the latter time period corresponds to the period of late circuitry maturation predicted in this study (P35-P56). This suggests that different insults (combined with different time periods) may affect different biological processes during PFC maturation, which may lead to different behavioral phenotypes.
Recently, the prodromal period of schizophrenia has been identified, in which impaired brain circuitry and functions are present before the onset of the disease [72,73]. By identifying critical changes in the brain during the prodromal period, we could develop early interventions to prevent the onset of the disorders, i.e., psychosis. Longitudinal studies using brain imaging showed that populations at high risk for schizophrenia developed progressive brain volume loss over time [74,75,76], which may be used as indicators for early interventions in these populations. There are numerous studies using brain imaging, trying to pinpoint the biological nature of these events [e.g., [77]]. It would be necessary to characterize both preclinical and clinical models in parallel [17], and by characterizing the animal models along the developmental trajectory of the PFC described here, we should be able to clarify the molecular signatures of prodromal phenotypes that may be translated into humans as useful biomarkers.
Conclusion
We have provided basic information of molecular profiles in the developing mouse PFC that may correlate with the maturation level of the brain region. Compared with similar information identified in primates [e.g., [78,79]], the results provided here in mice should be useful when we characterize the PFC of mouse models for developmental neuropsychiatric disorders in order to understand pathophysiology of the disorders as well as the biological nature of the prodromal state of schizophrenia.
Acknowledgements
Our work, performed at the Open Innovation Laboratory for Drug Discovery and Development by Takeda and Kyoto University's ‘Basic and Clinical Research Project for CNS Drugs', was supported in part by Takeda Pharmaceutical Co. Ltd. In addition, A.S. and M.N. are supported by the NIH (MH-084018, MH-094268 Silvio O. Conte Center, MH-069853, MH-085226, MH-088753, and MH-092443 to A.S.; K99MH-094408 to M.N.) and T.S., H.H., J.S., and T.K. are supported by grants from the Ministry of Education, Culture, Sports, Science and Technology in Japan/Japan Society for the Promotion of Science Grants-in-Aid for Scientific Research (25116516 and 25350987 to T.S.; 25123709, 22110007, and 24500408 to H.H.; 13J01992 to J.S.; 23115101 and 25250006 to T.K.). J.S. is a Research Fellow of the Japan Society for the Promotion of Science. We thank the Medical Research Support Center at Kyoto University Graduate School of Medicine for the fluorescence microscopy (supported by Platform for Drug Discovery, Informatics, and Structural Life Science from the Ministry of Education, Culture, Sports, Science and Technology in Japan) and Drs. Hikida and Tomoda of the Medical Innovation Center for critical reading and comments.
Disclosure Statement
The authors declare that there are no conflicts of interest regarding this article.
