

Abstract
Background: Drug combination therapies using cisplatin and natural products are common practice in the treatment of human lung cancer. Osthole is a natural compound extracted from a number of medicinal plants and has been shown to exert strong anticancer activities with low toxicity. Methods: In the present study, NBM-T-BMX-OS01 (BMX), derived from the semi-synthesis of osthole, was evaluated in cisplatin treated A549 cells to investigate its effect on cisplatin resistance in human lung cancer. The anticancer effect of BMX were measured by cell viablity‚ colony formation‚ TUNEL staining‚ flow cytometry and cell cycle assay. The fluorescence staining was performed to detect intracellular and mitochondrial reactive oxygen species (ROS) generation. Western blot analysis, antagonists pretreatment and small interfering RNA (siRNA) transfection were used to determine the potential mechanism. Results: It was found that, in comparison with single cisplatin treatment, the combination of BMX and cisplatin resulted in greater efficacy in inhibition of proliferation and colony formation, apoptosis induction and cell cycle arrest. The results of fluorescence staining showed that the combination effect of BMX and cisplatin was due to oxidative stress induced by mitochondrial ROS generation. In addition, BMX significantly attenuated the phosphorylation of ERK and Akt, two important pro-survival kinases. In contrast, BMX inhibited the activation of AMPK, and knockdown of AMPK using specific siRNA partially reversed BMX-induced inhibition of ERK and Akt, as well as its synthetic effects on cisplatin induced anticancer activity in A549 cells. Conclusion: Taken together, this study provides that BMX might modulate cisplatin resistance through AMPK-ERK and AMPK-Akt pathways. These results also support the role of BMX as a potential drug candidate for use in combination with cisplatin in the treatment of human lung cancer.
Introduction
Lung cancer is the most commonly diagnosed cancer in the western countries, and is considered to be the leading cause of cancer death in men and the second leading cause in women [1]. The prognosis of lung cancer patients is poor and the 5 year survival rate is still lower than 15% in spite of several therapeutic improvements achieved in recent years [2]. Non-small cell lung cancer (NSCLC), the most common histological type of lung cancers, accounts for approximately 75-80% of all lung cancers and causes more than one million deaths in the world each year [3]. More than 40% of NSCLCs are often found in an advanced stage, when it is too late for surgical intervention. Thus, chemotherapy with drugs such as platinum remains the main method of treating NSCLCs patients [4,5].
Although NSCLC is one of the most challenging cancers to treat, several previous clinical trials have demonstrated that systemic combination chemotherapy with platinum-based agents has advantages in prolonging survival and improving quality of life in NSCLC patients [6]. Cisplatin is the first platinum-based anti-cancer drug introduced to clinical treatment, and plays a central role in therapy for NSCLC. Just like many other chemotherapeutic agents, cisplatin interacts with cellular nucleophiles to form inter- and intra-stand cross links of DNA, RNA and/or proteins, leading to cell cycle arrest and apoptosis [7,8]. However, because of increased multidrug resistance to chemotherapeutic agents found in most NSCLCs, as well as cisplatin induced side effects, cisplatin-based chemotherapy for NSCLC patients seems to have reached a plateau [9]. For this reason, to explore the molecular mechanisms of chemotherapeutic resistance and find new targeted agents to enhancing the sensitivity of NSCLCs cells to chemotherapeutic agents is a promising therapeutic approach.
Cnidium monnieri (L.) Cusson, a small annual plant called “She chuang” in China, is a well-known traditional Chinese medicine and has been administered to humans for the treatment of eczema, cutaneous and sexual dysfunction for many years [10,11]. Osthole, (7-methoxy-8-isopentenoxycoumarin, C15H16O3, 244.39 Da), is an active substance isolated from the fruit of Cnidium monnieri (L.) Cusson and is responsible for most of its therapeutic functions. Previous investigations have shown that osthole possesses various beneficial pharmacological activities including: anti-osteoporotic, anti-inflammatory, anti-allergic, anti-seizure, and anti-diabetic effects [12,13,14,15]. Moreover, accumulating evidence has shown that osthole exerts anti-cancer effects through inducing cell cycle arrest and apoptotic cell death [16,17,18]. Recent studies have also shown that osthole is able to inhibit the migration and invasion of cancer cells through suppression of MMP-2 and MMP-9 enzyme activities [19,20]. However, the exact molecular mechanism of osthole induced anticancer effect is not completely understood, and there are no results to date of the effect of osthole on cisplatin-related chemotherapeutic resistance in human lung cancer cells. The aim of the present study therefore was to determine whether NBM-T-BMX-OS01 (BMX), a derivative semi-synthesized from osthole, would be effective in sensitizing human lung cancer A549 cells to cisplatin. We also investigated the potential molecular mechanism with focus on AMPK-mediated inhibition of ERK and Akt pathways.
Materials and Methods
Cell cultures
Human lung cancer A549 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum and 80 U/ml antibiotics (penicillin and streptomycin). The cells were maintained at 37°C in a humidified atmosphere of 95% air and 5% CO2, and the culture medium was changed every other day. NBM-T-L-BMX-OS01 (BMX) was dissolved in 0.1% dimethyl sulfoxide (DMSO), which had no effect on cell viability.
MTT assay
Cell viability was measured using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay, which assessed the ability of cells to convert MTT into the blue formazan dye [21]. Briefly, A549 cells were seeded in 96-well plates and subjected to various treatments as described earlier. MTT solution (5 mg/ml) was added to each culture well, and the cells were incubated for 4 h at 37°C. After the medium was carefully removed, the blue-colored formazan product was dissolved with DMSO and the absorbance was subsequently measured at 570 nm using a microplate reader. Cell survival rates were expressed as percentage of the value of cells without any treatment.
Colony Formation Assay
Cell survival after cisplatin and/or BMX treatment was detected by colony formation assay. A549 cells were incubated after cisplatin and/or BMX treatment for 14 days and then fixed with methanol and stained with 0.25% crystal violet. Colonies containing >30 cells were counted under a dissecting microscope. The results are reported as a percentage of the colonies in untreated cultures of each corresponding clone.
TUNEL staining
To detect apoptotic cell death in A549 cells, TUNEL staining was performed using an in situ cell death detection kit. Briefly, A549 cells were fixed by immersing slides in freshly prepared 4% methanol-free formaldehyde solution in phosphate buffered saline (PBS) for 20 min at room temperature. The cells were then permeabilized with 0.2% Triton X-100 for 5 min. Cells were labeled with fluorescein TUNEL reagent mixture for 60 min at 37°C according to the manufacturer's suggested protocol (Promega, Madison, WI, USA). Subsequently, the slides were examined by fluorescence microscopy and the number of TUNEL positive (apoptotic) cells was counted. The diamidino-phenyl-indole (DAPI, 10 µg/ml) was used to stain the nucleus.
Caspase-3 activity
The activity of caspase-3 was measured using a colorimetric assay kit according to the manufacturer's instructions (Cell Signaling, MA, USA). Briefly, after being harvested and lysed 106 cells were mixed with 32 µl of assay buffer and 2 µl of 10 mM Ac-DEVD-pNA substrate. Absorbance at 405 nm was measured after incubation at 37°C for 4 h. Absorbance of each sample was determined by subtraction of the mean absorbance of the blank and corrected by the protein concentration of the cell lysate. The results were described as relative activity to that of control group.
Flow cytometry
A549 cells were harvested 24 h after exposure to cisplatin and/or BMX, washed with ice-cold Ca2+ free PBS, and re-suspended in binding buffer. Cell suspension was transferred into a tube and double-stained for 15 min with the Alexa Fluor 488-conjugated annexin V (AV) and propidium iodide (PI) at room temperature in the dark. After addition of 400 µl binding buffer, the stained cells were analyzed by an FC500 flow cytometer with the fluorescence emission at 530nm and >575 nm. The CXP cell quest software (Beckman-Coulter, USA) was used to count the number of apoptotic (AV+/PI- and AV+/PI+) and necrotic (AV-/PI+) cells, and analyzed the results.
Assessment of the cell cycle
After various treatments, cells were washed, fixed in citrate buffer, and finally incubated for 1 h at -20°C. Cells were next incubated in a glycine/NaCl buffer containing 0.1% NonidetP-40,10 µg/ml RNase A, and 40 µg/ml of propidium iodide (PI) for 1 h at 4°C. Cell distribution across the different phases of the cell cycle was detected with a FACScan and analyzed.
ROS generation assay
Intracellular reactive oxygen species (ROS) generation were evaluated by determining the level of hydrogen peroxide (H2O2) using the probe 2',7'-dichlorofluorescin diacetate (DCFH-DA, Sigma). This probe was accumulated by cells and hydrolysed by cytoplasmic esterases to become 2',7'-dichlorofluorescin (DCFH), which were reacted with ROS to give the fluorescent product 2',7'-dichlorofluorescein (DCF). A549 cells were incubated with 50 µg/ml DCFH-DA for 1 h at 37°C in dark, and then re-suspended in PBS. Fluorescence was read using an excitation wavelength of 480 nm and an emission wavelength of 530 nm with a fluorescence plate reader.
Mitochondrial respiratory chain complex I activity
Mitochondria were purified by Percoll density gradient centrifugation in extraction buffer (50 mM Tris HCl, pH 7.5, 500 mM NaCl, 0.03 % reduced Triton X-100, 1 mM EDTA, 1 mM PMSF, 0.5 mM benzamidine, and 1 mg/ml each of pepstatin-A, leupeptin and aprotinin). All the samples were subjected to three freeze-thaw cycles to disrupt membranes and expose enzymes before analysis. The enzymatic activity of mitochondrial respiratory chain complex I was measured at 37°C as previously described [22]. The data were expressed as the percentage of control.
MitoSOX Assay
Mitochondrial superoxide production was measured by using the MitoSOX Red Kit (Invitrogen, Carlsbad, CA, USA). Briefly, A549 cells were incubated with MitoSOX Red for 10 min in a CO2 incubator at 37°C. MitoSOX was added to the medium to a final concentration of 5 µM after treatment and cells were rinsed with the perfusion buffer before imaging. Following incubation, MitoSOX Red fluorescence intensity was acquired at 510/580 nm on an Olympus FV10i Confocal Microscopy equipped with a digital cooled charged-coupled device camera.
ADP/ATP ratio assay
ADP/ATP ratios were determined using the Enzylight ADP/ATP ratio assay kit (BioAssay, Hayward, CA, USA), according to the manufacturer's instructions.
Small interfering RNA (siRNA) transfection
AMPK specific siRNA (sc-29673, Santa Cruz, CA, USA) and control siRNA (sc-37007, Santa Cruz, CA, USA) were dissolved separately in Optimem I (Invitrogen, CA, USA). After 10 min of equilibration at room temperature, each RNA solution was combined with the respective volume of the Lipofectamine 2000 solution (Invitrogen, CA, USA), mixed gently and allowed to form siRNA liposomes for 20 min. The A549 cells were transfected with the transfection mixture in antibiotic-free cell culture medium for 72 h, and subjected to various measurements.
Western blot analysis
Equivalent amounts of protein (40 µg per lane) were loaded and separated by 10% SDS-PAGE gels, and transferred to polyvinylidene difluoride (PVDF) membranes. Membranes were blocked with 5% nonfat milk solution in tris-buffered saline with 0.1% Triton X-100 (TBST) for 1 h, and then incubated overnight at 4°C with the primary antibody dilutions in TBST. The following antibodies were used: p-AMPK (2535), AMPK (2532), p-ERK (9101), ERK (9102), p-Akt (9271) and Akt (9272) obtained from Cell Signaling Technology. After that the membranes were washed and incubated with secondary antibody for 1 h at room temperature. Immunoreactivity was detected with Super Signal West Pico Chemiluminescent Substrate (Thermo Scientific, Rockford, IL, USA). An analysis software named Image J (Scion Corporation) was used to quantify the optical density of each band.
Statistical analysis
Statistical analysis was performed using SPSS 16.0, a statistical software package. Statistical evaluation of the data was performed by one-way analysis of variance (ANOVA). A value of P < 0.05 was considered statistically significant.
Results
BMX enhances cisplatin-induced anti-cancer effects in A549 cells
To confirm the anti-proliferative effect of BMX, human lung cancer A549 cells were treated with BMX at different concentrations, and the cell viability was assayed 24 h later. The results showed that BMX at concentrations higher than 5 µM exerted anti-cancer effects in A549 cells, but in contrast, no obvious toxicity was observed in BMX treated normal bronchial cell line HBE cells (Fig. 1A). To determine whether BMX altered sensitivity to cisplatin, A549 cells were treated with BMX before exposure to 10 µM cisplatin, and the results showed that BMX (even at concentration of 0.5 µM) significantly decreased cell viability compared to cisplatin treatment alone (Fig. 1B). In addition, 1 µM BMX was found to enhance cisplatin-induced decrease in cell viability in a time dependent manner up to 72 h after treatment (Fig. 1C). A similar result in colony forming assay was also observed as shown in Fig. 1D.
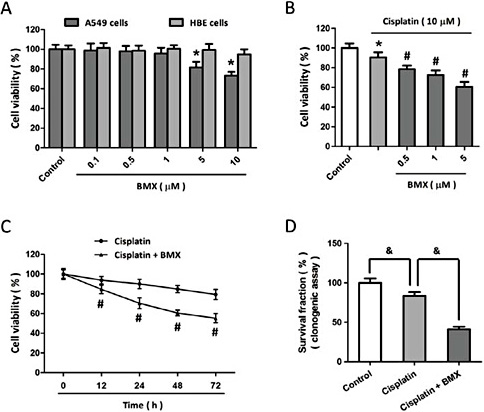
BMX enhances cisplatin-induced anti-cancer effects in A549 cells. The NSCLC A549 cells and normal bronchial cell line HBE were treated with the indicated concentrations of BMX (0.1, 0.5, 1, 5 or 10 µM), and the cell viability was measured by the MTT assay 24 h later (A). A549 cells were treated with 10 µM cisplatin with or without BMX at different concentrations (0.5,1 or 5 µM) for 24 h, and the cell viability was measured by MTT assay (B). A549 cells were treated with 10 µM cisplatin with or without 1 µM BMX. The cell viability was measured at different time points (C), and the mean clonogenic survival of cells were assayed at 14 d later (D). The data was represented as means ± SEM from five experiments. *p < 0.05 vs. control. #p < 0.05 vs. cisplatin. &p<0.05.
BMX enhances cisplatin-induced anti-cancer effects in A549 cells. The NSCLC A549 cells and normal bronchial cell line HBE were treated with the indicated concentrations of BMX (0.1, 0.5, 1, 5 or 10 µM), and the cell viability was measured by the MTT assay 24 h later (A). A549 cells were treated with 10 µM cisplatin with or without BMX at different concentrations (0.5,1 or 5 µM) for 24 h, and the cell viability was measured by MTT assay (B). A549 cells were treated with 10 µM cisplatin with or without 1 µM BMX. The cell viability was measured at different time points (C), and the mean clonogenic survival of cells were assayed at 14 d later (D). The data was represented as means ± SEM from five experiments. *p < 0.05 vs. control. #p < 0.05 vs. cisplatin. &p<0.05.
BMX increases cisplatin-induced apoptosis in A549 cells
To investigate the effect of BMX in cisplatin-induced apoptosis, A549 cells were treated with 10 µM cisplatin with or without 1 µM BMX, and the apoptotic cell death was detected by TUNEL staining (Fig. 2A). The results showed that cisplatin increased the number of TUNEL positive cells, which was further increased by BMX treatment (Fig. 2B). We also observed that BMX significantly enhanced cisplatin induced activation of caspase-3 in A549 cells (Fig. 2C). Furthermore, apoptosis and necrosis in A549 cells after cisplatin and/or BMX treatment were assayed by flow cytometry (Fig. 2D). As shown in Fig. 2E, BMX markedly increased cisplatin induced increase in apoptotic rate, with no effect on necrotic rate in A549 cells. The percentage of cells in individual cell-cycle phases was assessed after cisplatin and/or BMX treatment. Representative histograms at 24 h are shown in Fig. 2F. In BMX treated group, a significant fraction of A549 cells accumulated in the subG1 phase together with a drastic reduction of the percentage of cells in the G2/M phase of the cell cycle.

BMX increases cisplatin-induced apoptosis in A549 cells. A549 cells were treated with 10 µM cisplatin with or without 1 µM BMX. The apoptotic cell death was detected by TUNEL staining (A), and the number of TUNEL positive cells was calculated (B). The activity of caspase-3 was measured by a colorimetric assay kit (C). The apoptosis and necrosis were also measured by flow cytometery (D) and calculated (E). The percentage of cells in individual cell-cycle phases was assessed at 24 h later (F). The data was represented as means ± SEM from five experiments. *p < 0.05 vs. control and #p < 0.05 vs. cisplatin.
BMX increases cisplatin-induced apoptosis in A549 cells. A549 cells were treated with 10 µM cisplatin with or without 1 µM BMX. The apoptotic cell death was detected by TUNEL staining (A), and the number of TUNEL positive cells was calculated (B). The activity of caspase-3 was measured by a colorimetric assay kit (C). The apoptosis and necrosis were also measured by flow cytometery (D) and calculated (E). The percentage of cells in individual cell-cycle phases was assessed at 24 h later (F). The data was represented as means ± SEM from five experiments. *p < 0.05 vs. control and #p < 0.05 vs. cisplatin.
BMX increases cisplatin-induced ROS generation in A549 cells
To determine whether BMX affects the generation of intracellular ROS, A549 cells were treated with 10 µM cisplatin with or without 1 µM BMX. As shown in Fig. 3A, the intracellular ROS production was measured by H2DCFDA staining. The results showed that the cisplatin-induced generation of ROS was significantly enhanced by BMX treatment. The cisplatin-induced loss of mitochondrial complex I activity was enlarged by BMX (Fig. 3B). In addition, MitoSOX was used to specifically quantify the formation of mitochondrial ROS in A549 cells (Fig. 3C). Confocal microscopic imaging demonstrated a prominent enhancement in green fluorescence signal in A549 cells treated with 10 µM cisplatin, indicating an increase in superoxide levels in mitochondria (Fig. 3D). The cisplatin-induced increase in mitochondrial superoxide was further increased by BMX treatment, as indicated by a significant increase in mean intensity of fluorescence.
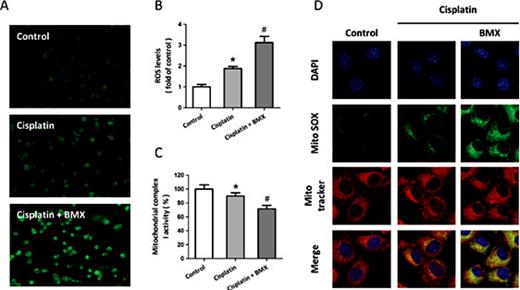
BMX increases cisplatin-induced ROS generation in A549 cells. A549 cells were treated with 10 µM cisplatin with or without 1 µM BMX. ROS generation was measured by DCF-DA staining (A), and the intracellular ROS levels cells in each group was calculated (B). The activity of mitochondrial complex I was measured (C), and the intra-mitochondrial ROS generation was assessed b< MitoSOX staining (D). The data was represented as means ± SEM from five experiments. *p < 0.05 vs. control and #p < 0.05 vs. cisplatin.
BMX increases cisplatin-induced ROS generation in A549 cells. A549 cells were treated with 10 µM cisplatin with or without 1 µM BMX. ROS generation was measured by DCF-DA staining (A), and the intracellular ROS levels cells in each group was calculated (B). The activity of mitochondrial complex I was measured (C), and the intra-mitochondrial ROS generation was assessed b< MitoSOX staining (D). The data was represented as means ± SEM from five experiments. *p < 0.05 vs. control and #p < 0.05 vs. cisplatin.
Involvement of Akt and ERK activation in cisplatin resistance in A549 cells
ERK and Akt are two important pro-survival factors in cancer cells, and western blot analysis was performed to detect the activation of ERK and Akt after cisplatin exposure in A549 cells. As shown in Fig. 4A, ERK was activated by cisplatin treatment up to 24 h after treatment, as evidenced by increased phosphorylation of ERK, which was peaked at 6 h. In contrast, only a temporal activation of Akt at 3 h was observed in A549 cells, and the phosphorylation of Akt was suppressed at 12 and 24 h after cisplatin exposure (Fig. 4B). To further investigate the involvement of these two protein kinases in cisplatin resistance in A549 cells, activation of ERK and Akt were inhibited by pretreatment with PD98059 and LY294002 respectively. The results showed that inhibition of ERK and Akt activation both significantly sensitized A549 cells to cisplatin, as evidenced by further decreased cell viability (Fig. 4C).

Involvement of Akt and ERK activation in cisplatin resistance in A549 cells. A549 cells were treated with 10 µM cisplatin, and the expression of p-ERK, ERK (A), p-Akt and Akt (B) were detected by western blot analysis. A549 cells were treated with 10 µM cisplatin in the presence or absence of Akt inhibitor LY294002 and ERK inhibitor PD98059, and the cell viability was measured by MTT assay (C). The data was represented as means ± SEM from five experiments. *p < 0.05.
Involvement of Akt and ERK activation in cisplatin resistance in A549 cells. A549 cells were treated with 10 µM cisplatin, and the expression of p-ERK, ERK (A), p-Akt and Akt (B) were detected by western blot analysis. A549 cells were treated with 10 µM cisplatin in the presence or absence of Akt inhibitor LY294002 and ERK inhibitor PD98059, and the cell viability was measured by MTT assay (C). The data was represented as means ± SEM from five experiments. *p < 0.05.
BMX enhances cell sensitivity to cisplatin via ERK and Akt inhibition
To determine whether BMX sensitize A549 cells to cisplatin through inhibition of ERK and/or Akt activation, A549 cells were treated with 10 µM cisplatin with or without 1 µM BMX. Results from western blot revealed that BMX significantly decreased ERK activation induced by cisplatin, whereas BMX alone did not alter the expression of p-ERK and ERK in A549 cells (Fig. 5A). As shown in Fig. 5B, BMX reduced the phosphorylation of Akt both in the presence and absence of cisplatin exposure, with no effects on Akt expression. PD98059 and LY294002 were used to inhibit ERK and Akt activation, and cell viability was assayed to investigate the potential synergetic effects of BMX and these inhibitors (Fig. 5C). The results showed that the anti-proliferative effects induced by PD98059 and LY294002 were both significantly strengthened by BMX, indicating the involvement of ERK and Akt activation in BMX induced anti-cancer effect.

BMX enhances cell sensitivity to cisplatin via ERK and Akt inhibition. A549 cells were treated with 10 µM cisplatin with or without 1 µM BMX, and the expression of p-ERK, ERK (A), p-Akt and Akt (B) were detected by western blot analysis. A549 cells were treated with 10 µM cisplatin in the presence or absence of 1 µM BMX, Akt inhibitor LY294002 and ERK inhibitor PD98059, and the cell viability was measured by MTT assay (C). The data was represented as means ± SEM from five experiments. *p < 0.05 vs. control. #p < 0.05 vs. cisplatin. &p < 0.05.
BMX enhances cell sensitivity to cisplatin via ERK and Akt inhibition. A549 cells were treated with 10 µM cisplatin with or without 1 µM BMX, and the expression of p-ERK, ERK (A), p-Akt and Akt (B) were detected by western blot analysis. A549 cells were treated with 10 µM cisplatin in the presence or absence of 1 µM BMX, Akt inhibitor LY294002 and ERK inhibitor PD98059, and the cell viability was measured by MTT assay (C). The data was represented as means ± SEM from five experiments. *p < 0.05 vs. control. #p < 0.05 vs. cisplatin. &p < 0.05.
BMX leads to inhibition of ERK and Akt through AMPK
We further investigated whether AMPK is involved in BMX induced regulation of ERK and Akt activation. As shown in Fig. 6A, treatment with BMX stimulated phosphorylation of AMPK even in the presence of cisplatin. Depleted ATP levels under conditions of energy deprivation were shown to trigger AMPK activation. Thus we measured ADP/ATP ratio to detect energy metabolism (Fig. 6B), and a marked increase in the cellular ADP/ATP ratio was observed after BMX treatment, indicating a decrease in the ATP content in A549 cells. After transfection with AMPK specific siRNA (Si-AMPK) or control siRNA (Si-control) for 72 h, A549 cells were treated with cisplatin and BMX. The results of western blot analysis showed that BMX induced inhibition on ERK (Fig. 6C) and Akt (Fig. 6D) activation were partially reversed by AMPK knockdown compared to Si-control transfected cells.

BMX leads to inhibition of ERK and Akt through AMPK. A549 cells were treated with 10 µM cisplatin with or without 1 µM BMX, and the expression of p-AMPK and AMPK were detected by western blot analysis (A). The ADP/ATP ratios were determined using the Enzylight ADP/ATP ratio assay kit (B). A549 cells were treated with 10 µM cisplatin in the presence or absence of transfection with AMPK siRNA (Si-AMPK) or control siRNA (Si-control), and and the expression of p-ERK, ERK (C), p-Akt and Akt (D) were detected by western blot analysis. The data was represented as means ± SEM from five experiments. *p < 0.05 vs. control. #p < 0.05 vs. cisplatin. &p < 0.05. vs. Si-control.
BMX leads to inhibition of ERK and Akt through AMPK. A549 cells were treated with 10 µM cisplatin with or without 1 µM BMX, and the expression of p-AMPK and AMPK were detected by western blot analysis (A). The ADP/ATP ratios were determined using the Enzylight ADP/ATP ratio assay kit (B). A549 cells were treated with 10 µM cisplatin in the presence or absence of transfection with AMPK siRNA (Si-AMPK) or control siRNA (Si-control), and and the expression of p-ERK, ERK (C), p-Akt and Akt (D) were detected by western blot analysis. The data was represented as means ± SEM from five experiments. *p < 0.05 vs. control. #p < 0.05 vs. cisplatin. &p < 0.05. vs. Si-control.
Involvement of AMPK in BMX-induced increase of cisplatin sensitivity
To further confirm the involvement of AMPK in BMX induced anti-cancer effects, A549 cells were treated with AMPK activator AICAR or transfected with Si-AMPK. The results showed that activation of AMPK using AICAR exerted anti-proliferative effects in A549 cells, as evidenced by decreased cell viability (Fig. 7A) and reduced survival fraction (Fig. 7B). The BMX induced decrease in cell viability was further decreased by AICAR, but partially reversed by Si-AMPK transfection (Fig. 7A). A similar result in colony forming assay was also observed (Fig. 7B).
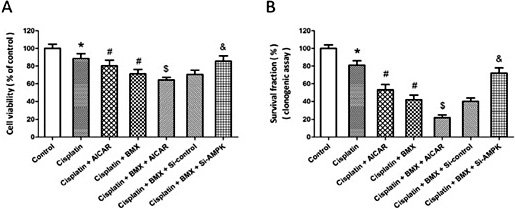
Involvement of AMPK in BMX-induced increase of cisplatin sensitivity. A549 cells were treated with 10 µM cisplatin in the presence or absence of 1 µM BMX, AMPK activator AICAR or transfection with AMPK siRNA (Si-AMPK) or control siRNA (Si-control). The cell viability was measured by MTT assay (A), and the mean clonogenic survival of cells were assayed at 14 d later (B). The data was re<resented as means ± SEM from five experiments. *p < 0.05 vs. control. #p < 0.05 vs. cisplatin. $p < 0.05 vs. cisplatin + BMX. &p < 0.05. vs. Si-control.
Involvement of AMPK in BMX-induced increase of cisplatin sensitivity. A549 cells were treated with 10 µM cisplatin in the presence or absence of 1 µM BMX, AMPK activator AICAR or transfection with AMPK siRNA (Si-AMPK) or control siRNA (Si-control). The cell viability was measured by MTT assay (A), and the mean clonogenic survival of cells were assayed at 14 d later (B). The data was re<resented as means ± SEM from five experiments. *p < 0.05 vs. control. #p < 0.05 vs. cisplatin. $p < 0.05 vs. cisplatin + BMX. &p < 0.05. vs. Si-control.
Discussion
Previous studies have demonstrated that osthole has potent anticancer activity in several human cancer cell lines [11,23], but the effect of osthole on cisplatin-related chemotherapeutic resistance has not been determined. In this report, we found that BMX, a derivative semi-synthesized from osthole, could sensitize human lung cancer A549 cells to cisplatin as evidenced by decreased cell proliferation and increased apoptosis. More extensive data at the molecular level led to the identification of an AMPK-dependent mechanism, which was related to the inhibition of two pro-survival signaling pathways: ERK and Akt. Our results not only confirmed the important roles of ERK and Akt activation in cisplatin-related chemotherapeutic resistance in NSCLC cells, which was consistent with other reports [24,25], but also demonstrated for the first time that osthole derivate exerted anticancer effects through AMPK-dependent inhibition of pro-survival pathways.
Cisplatin is the most commonly used active chemotherapeutic agent for the treatment of NSCLC, but its clinical use is limited due to severe side effects, including the acquisition of drug resistance [26,27]. In recent years, many investigations have focused on the combinatorial use of conventional chemotherapeutic agents and anticancer natural products to exert enhanced anticancer activity [28,29,30]. Osthole reportedly possesses anticancer effects by inhibiting cancer cell growth and metastasis in several human cancer cells, and its effects on cell migration and invasion have also been determined in human lung cancer cells [31,32]. As compared to many other anti-cancer agents, allicin represents unique advantages, such as high yields and low toxicity [33]. In the present study, the anti-proliferative activity of BMX, a derivative semi-synthesized from osthole, was also confirmed in A549 cells when it is administered at the concentration of 5 µM, but not in normal bronchial cell line HBE cells at all concentrations used. Intriguingly, even at the concentration of 0.5 µM, which is much lower than its effective concentration in lung cancer cells, BMX could still enhance the anti-proliferative effect of cisplatin. This combination treatment might be effective in decreasing the side effects of cisplatin since efficacy can be achieved with lower doses, which needs to be further investigated in further in vivo studies.
Free radicals are reactive compounds that have one or more unpaired electrons in their valence shell, and they are produced at low levels during normal physiological conditions and are scavenged by endogenous antioxidant systems [34]. ROS represent the most important class of radical species generated in cells, and ROS associated oxidative stress has been shown to play important roles in cancer [35,36,37]. ROS can promote carcinogenesis by playing a physiological role in intracellular signaling and regulation as secondary messengers. More importantly, ROS can act as anti-cancer agents via promoting cell-cycle arrest, apoptotic and necrotic cell death, and inhibiting angiogenesis [36,38]. Previous studies showed that cisplatin induced anti-cancer effects was partially mediated by ROS related oxidative, which was also demonstrated by fluorescent staining in our present study. There is growing evidence that destroyed oxidative stress scavenging system is involved in cisplatin resistance [8,27]. Increased glutathione (GSH) in cancer cells may cause resistance by binding/inactivating cisplatin, enhancing DNA repair, or reducing cisplatin-induced oxidative stress [39]. Over-activation of several GSH related enzymes, such as glutamate cysteine ligase, GSH reductase and catalase (CAT) have also been linked to cisplatin resistance [40,41]. In the present study, BMX was shown to increase intracellular ROS generation through promoting mitochondrial oxidative stress in A549 cells, which was accompanied by low efficacy in normal ATP supply. All these data indicated that BMX increased cisplatin-induced anti-cancer activity through targeting mitochondrial dysfunction and ROS associated oxidative stress.
The ERK and Akt signaling pathways consist of several kinases cascades that are regulated through phosphorylation and de-phosphorylation by specific kinases, phosphatases, adaptor proteins and scaffolding proteins [42]. These cascades play an important role in controlling cell proliferation, survival and invasion, and are considered to be main pro-survival pathways in living system [43,44]. Dysregulated signaling through ERK and Akt pathways is often the results of genetic alterations in critical components under disease conditions, and constitutive activation of these molecules is a common event in human cancer. In lung cancer patients, p-ERK and p-Akt were expressed at high levels, which were accompanied by increased expression of anti-apoptotic proteins Bcl-2, suggesting the involvement of these kinases in lung cancer cell survival [45]. Over-activation of ERK and Akt are reported to play a prognostic role and contribute to drug resistance in many cancers [46,47]. In the present study, we found that cisplatin treatment significantly increased ERK and Akt phosphorylation in time-dependent manners, and inhibitors of these kinases increased cisplatin induced anti-proliferative effects in A549 cells, which was consistent with a previous study [25]. Inhibition of Akt activation through PI3K inhibitors was shown to enhance cisplatin efficacy in resistant lines and ovarian cancer xenograft models [48,49]. An orally active difluorobenzamide PD-184352 was the first ERK inhibitor to undergo clinical testing, and it was demonstrated to achieve disease stabilization for more than 4 months in 25% patients [50]. Here, decreased activation of ERK and Akt was observed in BMX treated A549 cells, and BMX was shown to exert additive anti-cancer activity when treated together with ERK and Akt inhibitors, indicating the involvement of ERK and Akt inhibition in our experiments. The ERK and Akt pathways are regulated by several intra-pathway positive or negative feedback loops, and BMX might simultaneously regulated these two kinases through other tumor related genes, such as p53, which is reported to induce an undesired pro-proliferative effect via ERK and Akt activation [51].
AMPK is a sensor of energy status that plays an important role in cellular energy homeostasis [52]. It arose very early during eukaryotic evolution and has been shown to be involved in the control of cell growth, proliferation, autophagy, mitochondrial biogenesis and disposal [53,54]. Several viruses and cancer cells are found to establish mechanisms to down-regulate AMPK expression and activation, allowing them to escape its restraining influences on growth. AMPK activation was shown to be down-regulated in 90% of 350 cases of breast cancer patients [55], and the LKB1 associated regulation of AMPK was also observed in melanoma cells [56]. In human lung cancer patients, a positive p-AMPK expression was associated with increased overall survival and recurrence-free survival [57]. The anti-cancer effects were also demonstrated in the present study in A549 cells, and we found that AMPK activator further decreased cisplatin induced decrease in cell viability and colony formation. BMX induced sensitization to cisplatin was accompanied by increased expression of p-AMPK, indicating the involvement of AMPK phosphorylation in cisplatin resistance in lung cancer cells. A number of studies have revealed that ERK and Akt were two important downstream factors of AMPK signal pathway. The AMPK activator AICAR was found to reduce IGF-1 induced ERK activation [58], and AMPK activation by selenium down-regulated ERK activity, COX-2 and production of prostaglandin [59]. More recently, AMPK was shown to reverse the mesenchymal phenotype of cancer cells by targeting the Akt-MDM2-Foxo3a signaling axis [60]. In the present study, BMX induced inhibition of Akt and ERK was partially reversed by AMPK specific siRNA, suggesting that BMX induced sensitization to cisplatin in A549 cells might be mediated by AMPK-ERK and AMPK-Akt pathways.
Conclusions
In conclusion, the results from this study identified a major role of AMPK activation by BMX, a derivative semi-synthesized from osthole, in the attenuation of proliferation and colony formation in cisplatin-treated A549 cells through inhibition on ERK and Akt pathways. Furthermore, our study suggests that BMX should be developed as a pharmacological agent for use in combination with other anti-cancer drugs in the treatment of human lung cancer.
Disclosure Statement
The authors report no conflicts of interest.
Acknowledgements
This work was supported by a grant from the Scientific and Technological Research Project of Shaanxi Province (No. 2011K12-70).
References
T.-J. Chen and Y.-F. Zhou contributed equally to this work.

