

Abstract
Recent developments in genetic technology have given us the opportunity to look at diseases in a new and more detailed way. This Mini Review discusses monogenetic forms of childhood-onset primary osteoporosis, with the main focus on osteoporosis caused by mutations in WNT1 and PLS3, two of the most recently discovered genes underlying early-onset osteoporosis. The importance of WNT1 in the accrual and maintenance of bone mass through activation of canonical WNT signaling was recognized in 2013. WNT1 was shown to be a key ligand for the WNT-signaling pathway, which is of major importance in the regulation of bone formation. More recently, mutations in PLS3, located on the X chromosome, were shown to be the cause of X-linked childhood-onset primary osteoporosis affecting mainly males. The function of PLS3 in bone metabolism is still not completely understood, but it has been speculated to have an important role in mechanosensing by osteocytes and in matrix mineralization. In this new era of genetics, our knowledge on genetic causes of childhood-onset osteoporosis expands constantly. These discoveries bring new possibilities, but also new challenges. Guidelines are needed to implement this new genetic knowledge to clinical patient care and to guide genetic investigations in affected families.
Introduction
The term childhood-onset primary osteoporosis is used to describe a child with osteoporosis and no known underlying disease that can explain bone fragility [1]. Efforts have been made to decide upon diagnostic criteria, but reaching a univocal consensus has been difficult [2]. The most important feature of all types of osteoporosis is that the bone tissue has a compromised strength and therefore a propensity to fracture [3]. However, the underlying mechanisms for osteoporosis often differ between adult- and childhood-onset osteoporosis, with the latter more often having a direct genetic cause [4,5]. Osteoporosis secondary to severe disease, hormonal imbalances or abnormal nutritional status can affect patients of all ages, including children. This review focuses only on primary osteoporosis and regarding secondary osteoporosis, we refer to other recent reviews that address the subject [1,6].
Patients presenting with childhood-onset primary osteoporosis are often classified as having osteogenesis imperfecta (OI) or idiopathic juvenile osteoporosis (IJO) [7]. The diagnosis IJO in its strictest meaning differs from other forms of childhood-onset osteoporosis in that IJO often takes a benign course with a spontaneous resolution after the child reaches puberty. Furthermore, IJO often has a distinct debut in the prepubertal child with skeletal pain from the back, hips and feet [7,8]. However, not all cases of IJO have a favorable prognosis and, in some patients, the disease continues into adulthood and can result in lifelong disability [9]. OI is the term for a heterogeneous group of skeletal conditions, foremost characterized by susceptibility to fractures and is sometimes described as ‘brittle bone disease' [10]. This is a very inclusive description of a disease, which also explains the heterogeneity within the group of OI regarding both its clinical presentation and the involved underlying mechanisms. The term ‘childhood-onset primary osteoporosis' is even more inclusive and comprises all children with osteoporosis not secondary to other diseases. Childhood-onset primary osteoporosis includes IJO and OI, but also other forms of early-onset osteoporosis that sometimes are difficult to classify under any specific label. The diagnostic criteria for pediatric osteoporosis are presented in figure 1.
![Fig. 1. Diagnostic criteria for osteoporosis in children. In the absence of vertebral compression fractures, both of the two main criteria need to be fulfilled before the diagnosis of osteoporosis can be made in a child. In the presence of one or more vertebral compression fracture(s), the likelihood of osteoporosis is higher, and osteoporosis can be present without fulfillment of the two main criteria. Concerning the degree of trauma, there is no strict definition of what constitutes ‘mild-moderate trauma', but as a general rule, fractures after falls from above 3 m heights or accidents with motorized vehicles should not be considered as fragility fractures. Clinically significant fracture history as defined by the ISCD guidelines [2].](https://karger.silverchair-cdn.com/karger/content_public/journal/hrp/84/6/10.1159_000439566/2/m_000439566_f01.jpeg?Expires=1716293418&Signature=X0rROsotesmJpjSsB64a5J2--Zk0aVQpaQnFwVmLj6TBZEHNUmNvDxV9IVZu2LaFh7qeJFP3ErfF-wOxC6GqMjK0IO-9euFa65qRWwwL-WkH9w~-yoQC2ygcDx9JOZi8R0J6r-KUtma5bOAK1zwAdvUU-Bn4zCdNRpyrTpNGHuXvsLK768f7EWSxGQ05o16Q~RzeRmQ~GMCKyewvreFVd-OQ90W55A8rK17l9lWWvoQncUIEoiA5Ma6MSLoonSP-Ad0DqkkSxQYmYnfbexwm6naqsDdvU5oqL~BJl84OowVaKMd5DgIYf9vDghRLzWCbGJZ5ESTIkOWT8FVd3mEr3A__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Diagnostic criteria for osteoporosis in children. In the absence of vertebral compression fractures, both of the two main criteria need to be fulfilled before the diagnosis of osteoporosis can be made in a child. In the presence of one or more vertebral compression fracture(s), the likelihood of osteoporosis is higher, and osteoporosis can be present without fulfillment of the two main criteria. Concerning the degree of trauma, there is no strict definition of what constitutes ‘mild-moderate trauma', but as a general rule, fractures after falls from above 3 m heights or accidents with motorized vehicles should not be considered as fragility fractures. Clinically significant fracture history as defined by the ISCD guidelines [2].
Diagnostic criteria for osteoporosis in children. In the absence of vertebral compression fractures, both of the two main criteria need to be fulfilled before the diagnosis of osteoporosis can be made in a child. In the presence of one or more vertebral compression fracture(s), the likelihood of osteoporosis is higher, and osteoporosis can be present without fulfillment of the two main criteria. Concerning the degree of trauma, there is no strict definition of what constitutes ‘mild-moderate trauma', but as a general rule, fractures after falls from above 3 m heights or accidents with motorized vehicles should not be considered as fragility fractures. Clinically significant fracture history as defined by the ISCD guidelines [2].
Childhood-Onset Primary Osteoporosis: A Genetically Heterogeneous Group
Today, there are 16 different genes described in which mutations can cause a phenotype classified as OI [11]. Most of these genes code either for type I collagen or for proteins that are directly involved in the processing of type I collagen, which is the major protein in bone [12]. Although OI is genetically heterogeneous, the great majority (approximately 90%) of patients diagnosed with OI have dominant mutations in either COL1A1 or COL1A2, encoding pro-α1(I) and pro-α2(I) chains of type I collagen [13]. While dominant forms are the most common, autosomal recessive and X-linked forms of OI are also known.
In addition to these 16 genes causative of OI, defects related to the WNT signaling pathway can also result in childhood-onset osteoporosis. Biallelic loss-of-function mutations in the low-density lipoprotein receptor-related protein 5 gene (LRP5) were first described in osteoporosis-pseudoglioma syndrome, an autosomal recessive disease characterized by childhood-onset osteoporosis and blindness [14]. This was also the first finding suggesting that the WNT signaling pathway is of great importance in bone metabolism. LRP5 was known to be a co-receptor that together with another receptor, Frizzled (FZD), forms a complex necessary for canonical WNT signaling. The pathway's key role in bone mass accrual is now known, and it has been shown that heterozygous mutations, and less severe biallelic mutations, in LRP5 can give rise to a milder phenotype of early-onset osteoporosis without the eye phenotype [15]. Furthermore, genome-wide association studies have shown that common single nucleotide polymorphisms in LRP5 are associated with peak bone mass, bone mineral density (BMD) and fracture risk in the general population [16,17].
OI Classification
The classification of OI patients into different subgroups continues to be a debated issue. In 1849, Vrolik first coined the name osteogenesis imperfecta [18], and in 1979, Sillence et al. [19] proposed a classification of OI patients into 4 different groups (OI I-IV). The classification was based on inheritance pattern, clinical presentation and radiological findings and has since its publication been the accepted way to classify OI patients. However, along with the discovery of the various genetic causes of OI, every new gene discovered was given its own OI subgroup and number. WNT1 mutations are now listed in OMIM as a cause of OI type XV [20] and, especially for clinicians, this expansion of the OI subgroups has become confusing. In 2010, a revision was made concerning the nosology and classification of genetic skeletal disorders, and the committee recommended that the diagnosis and subclassification of OI should be made on phenotypic criteria and not on the exact molecular cause [21]. In 2014, Van Dijk and Sillence [11] proposed a classification of OI into 5 different groups (OI type I-V), together with descriptive names, again based on radiological and clinical characteristics (table 1). OI type V is characterized by very specific radiological findings, namely hyperplastic callus formation at fracture sites and calcification of the interosseous membrane of the forearm, and was therefore added to the original classification [22]. This revision of the OI classification is welcomed, especially by clinicians.

The characteristic traits most commonly associated with OI are skeletal fragility and extraskeletal features, including distinct blue hue of the sclerae and hearing impairment [23]. These characteristics are typical for patients with OI type I, which is also the group that comprises most of the OI patients. Interestingly, the first group of OI patients described by Ekman [24] in 1788 did not show these extraskeletal characteristics, again illustrating the broadness of the OI spectrum and the challenges in deciding on a uniform classifying system.
WNT1 and the WNT Signaling Pathway
WNT1(Wingless-Type MMTV Integration Site Family, Member 1) is one of the most recently discovered genes in which mutations can cause childhood-onset primary osteoporosis. WNT1 functions as a key ligand in the canonical WNT signaling pathway, which, as previously discussed, is one of the most important signaling pathways in bone regulation [25] (fig. 2). WNT1 promotes bone formation by binding to the LRP5-FZD receptor complex and thereby activates the canonical WNT signaling pathway [26].

The role of WNT1 in the WNT signaling pathway. In the mature bone, WNT1 signals in osteoblasts and osteocytes via the WNT signaling pathway. Signaling is initiated by binding of WNT1 to the receptor complex consisting of LRP5 and Frizzled. The binding of WNT1 activates an intracellular pathway that inhibits proteosomal degradation of β-catenin. Accumulating β-catenin translocates to the nucleus to control target gene expression that ultimately leads to increased bone formation and decreased bone resorption.
The role of WNT1 in the WNT signaling pathway. In the mature bone, WNT1 signals in osteoblasts and osteocytes via the WNT signaling pathway. Signaling is initiated by binding of WNT1 to the receptor complex consisting of LRP5 and Frizzled. The binding of WNT1 activates an intracellular pathway that inhibits proteosomal degradation of β-catenin. Accumulating β-catenin translocates to the nucleus to control target gene expression that ultimately leads to increased bone formation and decreased bone resorption.
WNT1 belongs to a family of 19 WNT proteins, many of which participate in fetal skeletal development and maintenance of postnatal bone health [26]. In the developing bone, WNT proteins ensure proper maturation and proliferation of bone cells by preventing the commitment of mesenchymal stem cells to chondrogenic and adipogenic lineages and by promoting their differentiation to osteoblasts and osteocytes [27,28]. WNT proteins also regulate osteoclastogenesis, both directly in an autocrine fashion and indirectly through osteoblasts [29]. However, WNT signaling is not only important for bone health, it is a signaling pathway known to regulate a wide range of developmental processes in various organs, especially during embryonic development. WNT proteins were first recognized for their role in oncogenesis, where aberrant WNT signaling can be linked to tumorigenesis and is implicated in many forms of cancers [30]. WNT signaling has also been shown to be involved in the development of the central nervous system. A murine mouse model, the swaying mouse, was known to exhibit severe cerebellar defects, which in 1991 was shown to be due to a homozygous WNT1 mutation [31]. The swaying mouse was described in 1967, but it was only after the first reports on WNT1 osteoporosis that its skeletal phenotype was characterized and shown to display significant osteoporosis [32].
WNT1 Osteoporosis
To date, primary osteoporosis due to WNT1 mutations has only been described in 10 families worldwide, the largest of these families involving 10 affected individuals [25]. WNT1 mutations leading to bone fragility include both homozygous autosomal recessive and heterozygous autosomal dominant mutations [25,33,34]. While much of the molecular and clinical characteristics of WNT1 osteoporosis remain to be solved, reviews of patient histories, clinical evaluations and imaging studies have given insight into the main characteristics of WNT1 osteoporosis.
All WNT1 mutations seem to lead to a reduction in bone mass as a consequence of decreased WNT signaling. The severity of osteoporosis varies greatly depending on the extent to which WNT1 signaling is affected. Laine et al. [25] described two siblings with severe skeletal phenotype with prenatal onset due to homozygous stop-gain mutations in WNT1. One of the children sustained her first fracture in utero, and also developed severe intellectual disability. Her MRI showed hypoplasia of the left cerebellar hemisphere, potentially related to the role of WNT1 in the development of the central nervous system. Both siblings showed severe long bone and spinal deformities and short stature. Other groups described similar findings in children with homozygous and compound heterozygous mutations in WNT1 [33,34,35]. Meanwhile, WNT1 osteoporosis caused by heterozygous loss-of-function mutations, resulting in less severely diminished WNT signaling, seem to result in milder forms of primary osteoporosis [25,33]. These patients are characterized by early-onset and progressive osteoporosis and multiple fractures, affecting mainly the spine, but with normal growth and only slightly shortened stature due to kyphosis. Other extraskeletal features, as well as abnormal biochemical findings, are usually not present [25].
Assessment of bone biopsies supports the role of WNT1 in osteoblast differentiation and function. Laine et al. [25] described histomorphometric findings in transiliac bone biopsies in 3 subjects with a heterozygous WNT1 mutation. All had significantly reduced bone turnover with low osteoblast and osteoclast numbers and activity. Keupp et al. [33] used XtremeCT to analyze 5 patients with heterozygous WNT1 mutations and could describe a reduction in relative bone volume, cortical thickness and trabecular density.
Treatment of WNT1 osteoporosis with bisphosphonates has been evaluated in a few patients, but remains to be further investigated. Palomo et al. [34] analyzed the effects of intravenous bisphosphonate treatment in 4 children with WNT1 osteoporosis. The authors described a slight increase in BMD and reshaping of compressed vertebrae as a result of treatment. However, the results did vary and were not as good as seen when treating patients with other forms of OI. Further studies on pharmacological treatment of WNT1 osteoporosis need to be conducted before any conclusions can be drawn.
PLS3: A Gene with Unknown Function
Mutations in PLS3 have recently been shown to be a cause of childhood-onset osteoporosis. Due to the gene's X-chromosomal location, PLS3 mutations affect male patients in a more severe manner than female patients [36]. The gene codes for the protein Plastin 3, an actin-binding and actin-bundling protein that is expressed in almost all solid tissues in the human body, and which is thought to be involved in cytoskeleton remodeling. Studies of the amino acid sequence of Plastin 3 identify 4 different domains: 2 calcium-binding and 2 actin-binding domains [37] (fig. 3). The 2 actin-binding domains can bind to separate actin filaments and, upon binding, the 2 actin filaments are cross-linked, forming bundles [38]. The actin-bundling properties of Plastin 3 have often been in focus, but experiments from Lyon et al. [39] suggest that the calcium-binding domains are the true effector domains and that Platstin 3 can function without interacting with actin filaments.
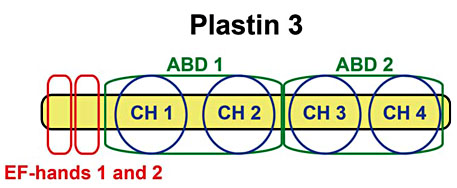
Structure of Plastin 3. The actin cytoskeleton is important for cell migration and adhesion, but also for controlling cellular shape and endo-/exocytosis. Plastin 3 is thought to be involved in the modification of the cytoskeleton via interaction with actin filaments. Upon binding of ABD1 and ABD2 to individual actin filaments, Plastin 3 can cross-link the filaments, forming actin bundles. By this action, Plastin 3 could potentially be involved in many cellular processes, but the function of Plastin 3 in bone is still unknown. EF-hands 1 and 2 are the two calcium-binding domains. ABD = Actin-binding domains; CH = calponin homology domain.
Structure of Plastin 3. The actin cytoskeleton is important for cell migration and adhesion, but also for controlling cellular shape and endo-/exocytosis. Plastin 3 is thought to be involved in the modification of the cytoskeleton via interaction with actin filaments. Upon binding of ABD1 and ABD2 to individual actin filaments, Plastin 3 can cross-link the filaments, forming actin bundles. By this action, Plastin 3 could potentially be involved in many cellular processes, but the function of Plastin 3 in bone is still unknown. EF-hands 1 and 2 are the two calcium-binding domains. ABD = Actin-binding domains; CH = calponin homology domain.
However, the function of PLS3 in bone is still unknown. Van Dijk et al. [36] suggested that Plastin 3 is involved in the process of mechanosensing, converting applied mechanical loading forces into molecular signals that are then interpreted by the cells. This suggestion is based on the observation that the chicken homologue of PLS3 is strongly expressed in the osteocytic dendrites, and it is the osteocytic dendrites that are thought to be most important for the osteocytes' mechanosensitivity. However, there are other suggestions for the function of Plastin 3 in the skeleton as well. Fahiminiya et al. [40] studied 4 boys with PLS3 mutations and childhood-onset osteoporosis. The authors evaluated transiliac bone biopsies from 2 of them and saw evidence of low osteoid maturation time, suggesting that Plastin 3 could be involved in the mineralization process. The authors also described a low amount of trabecular bone but with a normal lamellation pattern, which is in accordance with what Laine et al. [41] reported from bone biopsies of 5 patients with PLS3 mutations.
PLS3 Mutations: A Male Kind of Osteoporosis
While patients with mutations in PLS3 are characterized by low BMD, frequent peripheral fractures and vertebral compression fractures, they otherwise appear completely healthy. Biochemical measurements of affected patients have shown normal serum calcium and phosphate levels, urinary bone turnover markers within the normal range and no evidence of hypercalciuria [40,41]. Extraskeletal features that are often associated with OI such as blue sclerae, discolored teeth, joint hyperlaxity or short stature, are usually absent. To date, only 8 families with childhood-onset primary osteoporosis due to PLS3 mutations have been described, and the majority of the severely affected individuals are males [36,40,41]. Due to its location on the X chromosome, males have only one copy of PLS3, which explains why males can present with a more severe phenotype than females. Female patients also have a much more variable clinical presentation, as the female phenotypic spectrum ranges from mild osteopenia to symptomatic childhood-onset osteoporosis, resembling that of the male patients [41]. While several patients with osteoporosis due to PLS3 mutations have received bisphosphonate treatment and shown significant improvement in BMD, sometimes even with normalization of their BMD [36], it has also been reported that much of the gain in BMD is lost after discontinuation of the treatment [40].
Genetic Investigation of Childhood-Onset Osteoporosis
Childhood-onset osteoporosis can have many causes. In this review, we have focused on osteoporosis resulting from an inherited monogenic error, but secondary causes must always be excluded before a diagnosis of primary osteoporosis can be made. Children with recurrent fractures often have adverse dietary or lifestyle factors that need to be corrected before the diagnosis of primary osteoporosis can be made and detailed genetic analyses are justified [42]. A routine primary investigation, where the family history is of special importance, will shed light on possible causes. Transiliac bone biopsies can also be of help when investigating a child with osteoporosis, although interpretation of the results can be challenging and requires expertise [43]. Patients with various forms of OI have a fairly well-characterized histomorphometric profile [44]. Bone biopsies in patients with PLS3 and WNT1 mutations show low-turnover osteoporosis [25,41]. Bone biopsies also help to exclude osteomalacia and guide in treatment decisions [43]. Ifgenetic etiology is suspected based on these investigations, we recommend that the patient undergoes a genetic evaluation.
Suggestion for Genetic Screening
A complete and thorough examination of a patient with childhood-onset osteoporosis is helpful to determine the exact underlying genetic cause. However, at the individual level, the clinical presentation can vary greatly even within groups of patients with the same genetic mutation. Inheritance patterns can be of help in the genetic investigation, but in practice, it is almost impossible to guess which gene is damaged in each specific case. Instead, we believe that it is wise to make use of the technology at hand and search with a wide screen at an early stage (fig. 4). As about 90% of all patients with OI have mutations in COL1A1 or COL1A2, we recommend the screening of these 2 genes first. If no mutations can be found in these genes and the suspicion of a genetic etiology is strong, a wide genetic screening is further recommended. One option is to use a gene panel, which, in parallel, can sequence all known genes underlying an OI-like phenotype. A gene panel can be constructed by designing a specific capture kit that only targets the genes of interest, and then only the captured DNA library is sequenced. Another possibility is to perform whole-exome sequencing or whole-genome sequencing and then run the sequencing data through a customized filter, where only mutations of known clinical importance pass through. However, creating a smooth workflow concerning the genetic investigation for these patients can only be done at centers specialized in genetic investigations. Not only is the expertise in interpreting the results needed, but also an infrastructure for both computational resources and sequencing facilities is required. At least patients with a clear family history of early-onset osteoporosis would benefit from a thorough genetic investigation, as identification of the defective gene can help to establish long-term prognosis, enable accurate genetic counseling and may influence treatment decisions.
![Fig. 4. Genetic evaluation of a child with primary osteoporosis. This flow chart represents a suggestion of how to conduct a genetic investigation in a child with primary osteoporosis, after a careful phenotyping has been performed. The two genes COL1A1 and COL1A2 should be screened first, and if no mutations in these genes can be identified and the suspicion of an underlying genetic cause is strong, we further recommend genetic screening for a number of genes related to OI [11,21,25,33,35,36,40,41,50,51,52,53,54,55,56,57,58,59,60,61,62,63]. However, this is a resource-demanding approach and therefore not applicable in all clinics.](https://karger.silverchair-cdn.com/karger/content_public/journal/hrp/84/6/10.1159_000439566/2/m_000439566_f04.jpeg?Expires=1716293418&Signature=08mf9dNhwQfBqjpF2WgoskegFwKF7rfoRQVJgDu76cF1A7i9urNMUtO67wUqwRkGiUmnoLdpfOIYobTpEH3O7QVPzePyj7fm9XrzDP1hJUwg1tSpOHDTDSa4mTZ8WcBkAbjAxMLp9sEaCSh9fVfwXKq~ZNZoe7XHBluRQyIBzORJLpdB3kolhkUE8e4dBlpjK6d6xtbQ1~-vE6jtEvceJPwtbSPA6QTemjrr-EjGn7DnSFzbQUDPGjqBl7IvErCRAGWqrji~s-Ubu9wfkf1mNyZHm0sZCy6i72DHi2xmOdvaO0mfxdx~r3gfuyTIiz902YF5nkp1q0eZlD2N~ogBRA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Genetic evaluation of a child with primary osteoporosis. This flow chart represents a suggestion of how to conduct a genetic investigation in a child with primary osteoporosis, after a careful phenotyping has been performed. The two genes COL1A1 and COL1A2 should be screened first, and if no mutations in these genes can be identified and the suspicion of an underlying genetic cause is strong, we further recommend genetic screening for a number of genes related to OI [11,21,25,33,35,36,40,41,50,51,52,53,54,55,56,57,58,59,60,61,62,63]. However, this is a resource-demanding approach and therefore not applicable in all clinics.
Genetic evaluation of a child with primary osteoporosis. This flow chart represents a suggestion of how to conduct a genetic investigation in a child with primary osteoporosis, after a careful phenotyping has been performed. The two genes COL1A1 and COL1A2 should be screened first, and if no mutations in these genes can be identified and the suspicion of an underlying genetic cause is strong, we further recommend genetic screening for a number of genes related to OI [11,21,25,33,35,36,40,41,50,51,52,53,54,55,56,57,58,59,60,61,62,63]. However, this is a resource-demanding approach and therefore not applicable in all clinics.
Treatment Possibilities
Today, the pharmacological treatment possibilities for children with primary osteoporosis are still limited, despite the increasing knowledge of the underlying molecular pathology. All patients with osteoporosis require advice regarding physical activity and optimal diet. Vitamin D supplements are often required to treat or prevent vitamin D deficiency [1]. The diet should preferably be the only source of calcium, but nevertheless, supplements are sometimes needed. As of yet, pharmacological treatment has almost exclusively consisted of bisphosphonate treatment, either intravenously or orally. These drugs have become a widely accepted treatment in children with primary osteoporosis, despite the small number of randomized studies that have evaluated the effects of bisphosphonates in children. However, the existing evidence suggests that this approach is justified, at least for certain patients [45,46,47].
Even if the treatment possibilities are still limited, this might change in the near future. The more we learn about the underlying mechanisms, the closer we are to the development of specific treatments that can target the exact molecular etiology of the disease. This also means that it will be increasingly more important to get the correct molecular diagnosis for each patient. An example of a new osteoporosis treatment under development is the anti-sclerostin antibody, which has shown very promising results so far and is expected to reach the market in a few years [48]. Sclerostin is a bone-derived protein that normally inhibits canonical WNT signaling in the adult bone. Sclerostin binds to LRP5, preventing the LRP5-FZD complex from initiating canonical WNT signaling. By blocking this inhibitor with an antibody, canonical WNT signaling is amplified and bone formation favored. The significant role of sclerostin in the regulation of bone formation was first discovered when defects in the sclerostin gene were shown to underlie two diseases with severe hyperostosis, namely sclerosteosis and van Buchem disease [26]. Sclerostin is specifically produced and secreted by mature osteocytes and can modulate WNT signaling in the surrounding bone cells via diffusion through the lacuno-canalicular network [26]. This osteocyte-specific expression profile is also the reason why sclerostin is such a favorable drug target. Anti-sclerostin antibodies have not been tested in a pediatric setting, but it would be appealing to think that the patients with childhood-onset primary osteoporosis due to decreased WNT signaling (e.g. LRP5 and WNT1 mutations) could benefit greatly from this treatment [49]. Based on these recent developments, it is reasonable to think that we are heading in the direction of personalized medicine - where specific molecular defects are treated with specific drugs.
Conclusions
In summary, childhood-onset primary osteoporosis is a term for a genetically heterogeneous group of patients, comprising all children with osteoporosis not secondary to other diseases. A complete genetic investigation of patients with this condition is recommended, at least for affected patients with a family history of early-onset osteoporosis. The pharmacological treatment options are still limited, and evidence of their efficacy and safety in children is scarce. However, during the last decade, we have seen a dramatic increase in the knowledge on the underlying mechanisms in childhood-onset primary osteoporosis, and this gives reason for optimism regarding improvements in the care of affected children.
Acknowledgements
Our research is financially supported by the European Society for Paediatric Endocrinology Research Unit, the Swedish Research Council, the Swedish Childhood Cancer Foundation, the Academy of Finland, the Sigrid Jusélius Foundation, the Folkhälsan Research Foundation, the Novo Nordisk Foundation, the Helsinki University Hospital research funds, and through the regional agreement on medical training and clinical research (ALF) between Stockholm County Council and Karolinska Institutet.
Disclosure Statement
All authors state that they have no conflicts of interest.
