

Abstract
Liver fibrosis results from many chronic injuries and often progresses to cirrhosis, liver failure, portal hypertension, and hepatocellular carcinoma. Liver transplantation is the only treatment available for patients with advanced stages of liver fibrosis. Therefore, new strategies for anti-fibrotic therapy are required. Various kinds of hepatocyte damage result in inflammation, which leads to the activation of hepatic stellate cells (HSCs), which are the major source of myofibroblasts in the liver. Myofibroblasts proliferate in response to various kinds of cytokines, chemokines, and growth factors and produce extracellular matrix proteins, which forms the fibrous scar. Myofibroblasts undergo apoptosis and inactivation when the underlying causative etiologies are cleared. Here we describe our current knowledge of targeting the steps in HSC activation as therapeutic target for liver fibrosis.
Introduction
Liver fibrosis is an ineffective wound-healing process of the liver in response to repeated and chronic injury from many etiologies, such as infectious diseases (e.g. viral hepatitis), metabolic derangements (non-alcoholic steatohepatitis), exposure to toxins (e.g. alcohol liver diseases), or autoimmune diseases (e.g. primary biliary cirrhosis, primary sclerosing cholangitis, and autoimmune hepatitis). The morphology characteristics of liver fibrosis are the deposition of extracellular matrix (ECM), which is produced by myofibroblasts. Myofibroblasts are absent from the healthy liver, accumulate in the injured liver, and serve as the principle effector cells of fibrogenesis.
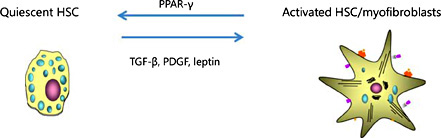
Several events are critical for pathogenesis of liver fibrosis and its resolution. Damage to hepatocytes cause inflammatory reactions, which lead to the activation of hepatic stellate cells (HSCs). HSCs in the normal liver are in a quiescent state and function to store retinoids and serve as the pericytes for the sinusoidal endothelial cells (fig. 1). Continuous liver injury causes perpetuation of activated HSCs, and they become myofibroblasts. Myofibroblasts proliferate in response to various cytokines, chemokines, and growth factors and produce ECM proteins. However, myofibroblasts may undergo apoptosis and inactivation when the underlying causative etiologies are removed (fig. 2). Control and clearance of the underlying causative etiology (e.g. virus suppression or alcohol absence) can slow down fibrosis progression and lead to fibrosis regression. However, our extensive knowledge on the mechanism leading to liver fibrosis through hepatocyte injury, inflammation, and activation of myofibroblasts to deposit ECM has not been translated into effective therapies in humans so far [1]. In this review, we summarize the current knowledge of targeting common pathways that lead to liver fibrosis.


Phenotypic pathway of HSCs. Hepatocyte damage causes an inflammatory reaction, which leads to the activation of HSCs. Continuous liver injury causes perpetuation of activated HSCs in the liver, and they become myofibroblasts. Myofibroblasts proliferate in response to various kinds of cytokines and produce ECM proteins. Myofibroblasts undergo apoptosis or inactivation when the underlying causative etiologies are cleared.
Phenotypic pathway of HSCs. Hepatocyte damage causes an inflammatory reaction, which leads to the activation of HSCs. Continuous liver injury causes perpetuation of activated HSCs in the liver, and they become myofibroblasts. Myofibroblasts proliferate in response to various kinds of cytokines and produce ECM proteins. Myofibroblasts undergo apoptosis or inactivation when the underlying causative etiologies are cleared.
Inhibition of Hepatic Injury
Liver injury is characterized by hepatocyte damage and death, increased inflammatory cells, and activated HSCs/myofibroblasts. Pharmacological inhibition of liver cell apoptosis may potentially attenuate liver injury, inflammation, and fibrosis by blocking hepatocyte death. Apoptosis is executed by a family of intracellular proteases referred to as caspases [2]. For example, a pan-caspase inhibitor IDN-6556 attenuated hepatic injury and fibrosis in mice [3]. Furthermore, ocadeic acid protects in part from liver fibrosis by protecting hepatocytes from injury [4].
Since reactive oxidative stress (ROS) mediates hepatocyte death, regulating ROS is a promising strategy of liver fibrosis therapy [5]. Peroxisome proliferator-activated receptor delta (PPARδ), a member of the nuclear receptor family, is a key metabolic regulator with pleiotropic actions on various tissues including fat, skeletal muscle, and liver. PPARδ agonist protects hepatocytes from cell death by reducing ROS generation of hepatocytes, leading to less liver fibrosis [6].
Nonalcoholic fatty liver disease (NAFLD) includes a spectrum of diseases ranging from isolated hepatic steatosis to nonalcoholic steatohepatitis (NASH), the progressive form of the disease associated with inflammation and cellular injury, which can lead to cirrhosis. NAFLD has become the most common chronic liver disease in the United States. It is associated with obesity, type 2 diabetes, hyperlipidemia, insulin resistance, and the accumulation of triglycerides in hepatocytes. Although the pathogenesis of the hepatocyte damage in response to lipid accumulation is not fully elucidated, cellular membrane integrity seems to be important for regulating hepatocyte damage. Phosphatidylcholine (PC) is a major component of the cellular membrane, which is generated by a transmethylation reaction from phosphatidylethanolamine through a metabolic pathway that utilizes S-adenosylmethionine (SAMe) as a methyl donor. The PC/PE ratio may be a key regulator of cell membrane integrity and play a role in the progression of steatosis to NASH. Animal studies show that chronic hepatic SAMe deficiency causes NASH and hepatocellular carcinoma (HCC). Furthermore, the formation of PC is reduced in various kinds of chronic liver diseases including intrahepatic cholestasis, cholestasis of pregnancy and alcoholic liver disease.
Inhibition of Inflammation
Serum amyloid P (SAP) or pentraxin-2, a member of the pentraxin family, is a 27-kDa protein that is produced by the liver, secreted into the blood, and circulates as stable135-kDa pentamers [7,8,9]. SAP reduces neutrophil adhesion to ECM proteins, inhibits the differentiation of monocytes into fibrocytes, decreases profibrotic macrophages, activates the complement pathway, and promotes phagocytosis of cell debris. SAP reduces bleomycin-induced lung fibrosis [10]. Injection of SAP into humans, mice and rats has no toxic effects.
Inhibition of Activation of HSCs
After stimulation by cytokines or engulfment of apoptotic bodies, Kupffer cells/macrophage are stimulated to produce TGF-β1 [11,12], which is a potent cytokine to activate HSCs into myofibroblasts with increased expression of α-SMA, TGF-β, platelet derived growth factor (PDGF), CTGF, type I collagen, and tissue inhibitor of metalloproteinase 1 (TIMP1) [13]. Although TGF-β1 is one of the most potent stimuli of ECM synthesis, suppressing its expression remains a major challenge of antifibrotic therapy, since systemic blocking of TGF-β1 can provoke inflammation and increase the risk of neoplasia. Neutralization of TGF-β in animal models inhibits liver fibrosis and reduces the risk of developing cholangiocarcinoma [14,15]. Fresolimumab (GC1008) is a human anti-TGF-β1 monoclonal antibody that neutralizes all isoforms of TGF-β. In patients with advanced malignant melanoma and renal cell carcinoma, fresolimumab demonstrated acceptable safety and preliminary evidence of antitumor activity [16,17,18]. Using radio-labeled (89) Zr-conjugated fresolimumab for PET to analyze TGF-β expression, GC1008 accumulated in primary tumors and metastases in a manner similar to IgG (89), and Zr-fresolimumab uptake is seen in sites of tumor ulceration and in scar tissue, where TGF-β is highly active [19]. Although there is a phase II clinical trial ongoing of fresolimumab, optimal strategies are still needed to restrict it to the fibrotic milieu. TGF-β transduces its signal to target genes through the ALK5 Ser/Thr kinase receptor. GW6604 (2-phenyl-4-(3-pyridin-2-yl-1H-pyrazol-4-yl) pyridine), an ALK5 inhibitor, inhibits the transcription and deposition of ECM and improves the deterioration of liver function in mice [20]. Yet, considering the pleiotropic effects of TGF-β, treatment with an ALK5 inhibitor should be carefully examined to avoid unwanted side effects [21].
Integrin αvβ1, which is absent in normal liver but expressed on activated HSCs, promotes liver fibrosis by activating latent TGF-β1. Selective deletion of integrin αv on HSCs inhibits liver fibrosis [22]. A small molecule inhibitor of αvβ1 prevents TGF-β1 activation and inhibits experimental liver fibrosis [23].
Lysophosphatidic acid (LPA) is a lipid mediator, which is produced mainly by activated platelets by the hydrolysis of lysophosphatidylcholine by autotaxin. LPA is a bioactive lipid implicated in several functions, including proliferation, apoptosis, migration, and cancer cell invasion [24]. LPA and LPA1 receptor (LPA1R) are increased in many inflammatory states, including pulmonary fibrosis, liver fibrosis, and systemic sclerosis [25]. LPA exerts various physiological effects on the receptors of parenchymal cells and LPA1R antagonists showed anti-fibrotic effect on models of liver fibrosis, lung fibrosis and scleroderma [25,26,27].
Inhibition of Proliferation of HSCs
Inhibitors of receptor tyrosine kinase and Ser/Thr kinase also demonstrate anti-fibrosis effects. The multitargeted receptor tyrosine kinase inhibitor sorafenib, which has been approved for the treatment of advanced renal cell carcinoma and HCC, and sunitinib can improve experimental hepatic fibrosis, inflammation, and angiogenesis [28,29]. SiRNA of transient receptor potential melastatin 7, a nonselective cation channel with protein serine/threonine kinase activity, attenuates TGF-β1-induced expression of myofibroblast markers, increases the ratio of matrix metalloproteinases (MMPs)/TIMPs, and decreases the phosphorylation of Smad2 and 3 associated collagen production [30,31]. Hepatic nuclear factor kappa B-inducing kinase, a Ser/Thr kinase, which is increased in injured livers in both, mice and humans, induces hepatocyte injury, activates bone marrow-derived macrophages, and leads to liver fibrosis, and this might serve as a target for liver fibrosis therapy [32].
The renin angiotensin pathway in HSCs induces reactive oxygen species and accelerates hepatic fibrosis [33]. In response to sustained liver injury, the renin angiotensin system (RAS) locally accelerates inflammation, tissue repair and fibrogenesis by production of angiotensin II (Ang II). RAS is a single cascade where renin converts angiotensinogen into angiotensin I, which in turn is converted to Ang II by angiotensin converting enzyme (ACE). Ang II mediates biological responses through 2 G-protein-coupled receptors, the Ang II receptor type 1 (AT1) and Ang II receptor type 2. However, the fibrogenic actions of Ang II are mostly mediated by angiotensin receptor AT1. Stimulation of AT1 receptor by Ang II results in the proliferation of HSCs and ECM deposition. Ang II also plays an important role in ROS formation by activating NADPH oxidase (NOX) in HSCs. In concordance, several experimental models of liver fibrosis in rodents have demonstrated that prolonged administration of Ans II directly causes HSC activation. Mice lacking AT1 receptors are protected from liver fibrosis. This makes RAS an attractive target for anti-fibrotic therapy. Although several small studies showed the usefulness of ACEi/ARB for liver fibrogenesis in patients with hepatitis C [34], the HALT-C cohort study did not show any anti-fibrogenic effects of ACEi/ARB for chronic hepatitis C patients [35].
Activation and proliferation of HSCs require NOX/ROS signaling. Chronic liver injury induces ongoing hepatocyte injury and death increases ROS production and decreases antioxidant activity, which is one of the characteristics of chronic liver disease that triggers liver fibrogenesis. NOX produces ROS by transferring electrons from nicotinamide adenine dinucleotide phosphate to molecular oxygen, which is different from other redox enzymes that produce superoxide as a byproduct. The mammalian NOX family is composed of 7 isoforms: NOX1, NOX2, NOX3, NOX4, NOX5, DUOX1, and DUOX2, which are distinctively expressed in specific cell types in the liver. HSCs express 3 NOX isoforms, NOX1, NOX2, and NOX4 [36,37] (fig. 3). NOX2 is the classic enzyme that phagocytic cells use to produce ROS to kill bacteria, and as such is not a good target for anti-fibrotic therapy. HSCs from p47phox-deficient mice (without a regulatory component of NOX) fail to generate ROS in response to Ang II, PDGF, leptin, or apoptotic bodies, and p47phox-deficient mice demonstrate reduced liver fibrosis after BDL or the hepatotoxin CCl4 [38,39]. NOX1 and NOX4 are expressed as activated HSCs, but only at very low levels in the uninjured liver. GKT137831, a potent dual NOX1/NOX4 inhibitor, attenuates ROS production and inhibits activation of HSCs [37] and experimental liver fibrosis [40]. Multicenter, long-term clinical trials are needed to evaluate the role of antioxidants in NASH.

Promotion of Apoptosis of Activated HSCs
Daily cannabis use is an independent risk factor for increased liver fibrosis in HCV patients [41]. CB1 and CB2 receptors are increased in liver fibrosis. CB1 KO mice are resistant to liver fibrosis, while CB2 KO mice have increased liver fibrosis [42,43]. CB1 agonists activate HSCs to myofibroblasts. CB1 receptor agonists, such as Rimonabant, inhibit and reverse experimental liver fibrosis [44,45]. A peripherally acting CB1 antagonist could treat liver fibrosis without inducing depression.
ECM degradation is mediated by (MMPs), a family of zinc-dependent enzymes grouped into collagenases, gelatinases, stromelysins, and membrane-type MMPs. MMP activity is regulated by TIMPs 1-4, which bind in substrate- and tissue-specific manners to MMPs, blocking their proteolytic activity. In concordance, monoclonal Anti-TIMP1 Ab partially reverses established CCl4-induced fibrosis [46]. Furthermore, persistent expression of TIMP-1 in vivo was associated with the persistence of activated HSCs, and during resolution of fibrosis, a decrease in TIMP-1 protein levels correlated with decreased numbers of HSCs [47].
Promotion of HSC Inactivation
Clinical and experimental hepatic fibrosis is reversible. Regression of liver fibrosis is associated with resorption of fibrous scar and disappearance of collagen producing myofibroblasts. The fate of these myofibroblasts has been recently revealed: some myofibroblasts undergo apoptosis during the regression of fibrosis, while other myofibroblasts revert to a quiescent-like phenotype. Inactivation of myofibroblasts is a newly described phenomenon [48] (fig. 4), which now requires mechanistic investigation. Inactivation of HSCs is associated with the re-expression of lipogenic genes PPAR-γ, Insig1, and CREBP. PPAR-γ is reported to be important for maintaining and for re-establishing the quiescent phenotype [49].
Inhibition of Deposition of Type I Collagen
In liver fibrosis, type I collagen is the most prominent increased components of the ECM. The cross-linking of type I collagen is also increased, which is modulated by the matrix enzyme lysyloxidase-like-2 (LOXL2). Although blocking collagens have unwanted off-target effects, the inhibition of LOXL2 by a monoclonal antibody (AB0023) reduces the production of cytokines, attenuates TGF-β signaling, and inhibits the activate fibroblasts (Barry-Hamilton, 2010). Similar to AB0023, another humanized monoclonal LOXL2 antibody (GS-6624) is in randomized, double blind, phase II clinical trials to treat NASH and PSC [50].
Conclusion
The cellular and molecular mechanism of liver fibrosis has been extensively studied, and new therapies based on these understandings are currently in clinical development. A list of potential therapies for liver fibrosis is provided in figure 5.

Disclosure Statement
No potential conflict of interest relevant to this article was reported.
Grant Support
NIH 2 P50 AA011999, 5 P42 ES010337, 5 U01 AA021856.
