

Abstract
Objective: The aim of this study was to elucidate any electrophysiological changes that may contribute to the development of neuropathic pain during treatment with the anticancer drug paclitaxel, particularly in the γ-aminobutyric acid (GABA) system. Materials and Methods: One hundred and eight Sprague-Dawley rats were used (untreated control: 43; vehicle-treated: 21, and paclitaxel-treated: 44). Paclitaxel (8 mg/kg) was administered intraperitoneally on 2 alternate days to induce mechanical allodynia. The rats were sacrificed 7 days after treatment to obtain slices of the anterior cingulate cortex (ACC), a brain region involved in the central processing of pain. Field excitatory postsynaptic potentials (fEPSPs) were recorded in layer II/III of ACC slices, and stimulus-response curves were constructed. The observed effects were pharmacologically characterized by bath application of GABA and appropriate drugs to the slices. Results: The paclitaxel-treated rats developed mechanical allodynia (i.e. reduced withdrawal threshold to mechanical stimuli). Slices from paclitaxel-treated rats produced a significantly higher maximal response (Emax) than those from untreated rats (p < 0.001). Bath application of GABA (0.4 µM) reversed this effect and returned the excitability to a level similar to control. Pretreatment of the slices with the GABAB receptor blocker CGP 55845 (50 µM) increased Emax in slices from untreated rats (p < 0.01) but not from paclitaxel-treated rats. Conclusion: In this study, there was a GABA deficit in paclitaxel-treated rats compared to untreated ones. Such a deficit could contribute to the pathophysiology of paclitaxel-induced neuropathic pain (PINP). Thus, the GABAergic system might be a potential therapeutic target for managing PINP.
Introduction
Paclitaxel is a chemotherapeutic agent whose use for the treatment of various types of solid tumors is hampered by a dose-limiting painful peripheral neuropathy [1]. Unfortunately, there are no clinically approved agents available to prevent the development of paclitaxel-induced neuropathic pain (PINP) or chemotherapy-induced neuropathic pain in general. For the treatment of chemotherapy-induced neuropathic pain, only duloxetine has proven efficacy [2]. Therefore, the need for further research into both the pathophysiology of PINP and the development of novel and efficacious agents for PINP treatment cannot be overemphasized.
The anterior cingulate cortex (ACC) is a prefrontal cortical region of the brain known to be involved in pain perception and modulation [3]. Imaging and neurophysiologic studies revealed increased activity in the ACC in patients and animal models of neuropathic pain [4,5], which could be due to a deficiency in γ;-aminobutyric acid (GABA)-ergic activity in this area [6,7]. The alterations in the GABAergic system play a role in the pathogenesis of neuropathic pain [8], and our group recently reported that dysregulation of the GABAergic system in the ACC possibly contributes to PINP [6].
Enaminone compounds, which are enamines of β-dicarbonyl compounds, such as the anilino enaminone methyl 4-(4′-bromophenyl)aminocyclohex-3-en-6-methyl-2-oxo-1-oate (E139), have anticonvulsive effects via enhancing extracellular GABA levels in addition to other mechanisms [9,10]. E139 may therefore have potential for use in treating PINP.
Since to our knowledge no studies have investigated electrophysiological alterations or the effects of GABA or E139 on fast synaptic transmission in the ACC during PINP, we examined them in the current study.
Materials and Methods
Animals
All animals (n = 108; 43 control, 21 vehicle-treated, and 44 paclitaxel-treated) used in this study were male Sprague-Dawley rats (200-300 g) obtained from the Kuwait University Health Sciences Animal Resource Center. The animals were handled in compliance with Directive 2010/63/EU of the European Parliament and of the Council on the protection of animals used for scientific purposes. All procedures were approved by the Ethical Committee for the use of Laboratory Animals in Teaching and Research, Health Sciences Centre, Kuwait University.
Chemicals and Drugs
Compound E139 was synthesized in house [11]; stock solutions were prepared in dimethyl sulfoxide (Sigma; St. Louis, Mo., USA), aliquoted into 40-μl portions, stored at -20°C, and used within 2 weeks. Paclitaxel and GABA were purchased from Tocris (Bristol, UK), CNQX from Tocris (Ellisville, Mo., USA); ethanol, Cremophor EL, CGP 55845, the salts used in the preparation of artificial cerebrospinal fluid (aCSF), and recording solutions were from Sigma (St. Louis, Mo., USA). All stock solutions were diluted to the desired concentrations with aCSF before use and applied to the tissue slices by bath perfusion.
Administration of Paclitaxel to Induce Neuropathic Pain
Paclitaxel was dissolved in a solution made up of 50% Cremophor EL and 50% absolute ethanol to a concentration of 6 mg/ml, stored at -22°C, and used within 2 weeks. This stock solution was diluted in normal saline (NaCl 0.9%) to a final concentration of 3 mg/ml just before administration. Paclitaxel (8 mg/kg body mass), or its vehicle, was administered to rats intraperitoneally in a volume of 2.67 ml/kg on 2 alternate days.
Assessment of Mechanical Allodynia
Mechanical allodynia in rats was measured using the dynamic plantar esthesiometer (Ugo Basile, Italy), as previously described [12,13]. Briefly, rats were left to habituate for about 15 min inside plastic enclosures on top of a perforated platform before measuring the withdrawal threshold (grams) to an automated filament that exerted a linearly increasing force (2.5 g/s with a cutoff time of 20 s) on the hind paw.
ACC Slice Preparation for Electrophysiological Recording
Briefly, rats were deeply anesthetized with halothane and decapitated; brains were quickly removed and immersed in ice-cold (4°C) aCSF bubbled continuously with 95% O2 and 5% CO2. A block of the forebrain containing the ACC was cut and glued to the cutting slab with cyanoacrylic glue and sectioned using a tissue slicer (Leica VT 1000S; Leica Microsystems, Wetzlar, Germany) into 350-µm-thick coronal slices. A slice was cut in half, trimmed, and placed in a 500-µl recording chamber where it was suspended on a nylon mesh and perfused with carbogenated aCSF at a flow rate of 2-3 ml/min. The recording bath temperature was maintained at 29-30°C throughout the course of each experiment using a heat circulator and exchanger. The composition of the aCSF used for dissection, storage, and recording was (in mM) NaCl (120), KCl (3.3), MgSO4 (1.2), CaCl2 (1.3), NaHPO4 (1.23), NaHCO3 (25), and D-glucose (10).
Field Excitatory Postsynaptic Potential Recording
For extracellular field recording, a glass recording electrode (3-6 MΩ) was pulled from a KG-33 glass, filled with 3 M NaCl, and placed in area II/III of the ACC. A bipolar tungsten stimulating electrode was placed in an area close to the recording electrode to activate afferent fibers with a chosen current produced by a stimulus isolator (AMPI-Isoflex, Manchester, UK), and the recording electrode was moved around until an optimal field excitatory postsynaptic potential (fEPSP) was recorded. The maximal stimulus was determined by applying a range of increasing stimulation strengths (0.5-5 V) until the fEPSP did not increase with higher stimulation intensities (saturated). A test stimulus was selected to be one that yielded between 40 and 50% of the maximum response recorded using the range of stimuli. Control (baseline) responses were recorded by delivering the test stimulus every 30 s (0.033 Hz), ensuring response stability for at least 10 min before any experimental manipulation. Ten randomly selected experiments were terminated with bath application of the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)/kainate (non-N-methyl-D-aspartate; non-NMDA) receptor blocker, 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX, 10 µM), to confirm that the fEPSPs were non-NMDA receptor mediated. All recordings were made using an Axopatch 1D amplifier and pClamp software (Clampex 10.0; Molecular Devices, Sunnyvale, Calif., USA) in current clamp mode at sampling rates of 50 kHz, filtered at 1 kHz, digitized, and stored for off-line analysis. Each stored fEPSP trace was an average of 2 consecutive fEPSPs elicited 30 s apart.
Statistical Analyses
All data are expressed as means ± SEM. Statistical analyses were performed using Student's t test (paired or unpaired), one-way analysis of variance (ANOVA) followed by Dunnett's multiple comparison test, or two-way repeated-measure ANOVA followed by Bonferroni post hoc tests. The differences were considered significant at p < 0.05.
Results
Paclitaxel-Induced Mechanical Allodynia in Rats
Treatment with paclitaxel significantly reduced the withdrawal threshold to mechanical stimuli (mechanical allodynia) on days 3-20 after the first drug administration (21.6 ± 0.9 to 38.7 ± 1.2 g) compared to the baseline mechanical threshold (42.9 ± 1.0 g) and vehicle-only-treated animals (p < 0.05; fig. 1).

Time course of the withdrawal threshold to the dynamic plantar esthesiometer of Sprague-Dawley rats after administration of 8 mg/kg paclitaxel or vehicle on 2 alternate days (n = 12-14 animals). * p < 0.05, ** p < 0.01, vs. vehicle on the same day after treatment; # p < 0.05, ## p < 0.01, vs. pretreatment (PRE) values.
Time course of the withdrawal threshold to the dynamic plantar esthesiometer of Sprague-Dawley rats after administration of 8 mg/kg paclitaxel or vehicle on 2 alternate days (n = 12-14 animals). * p < 0.05, ** p < 0.01, vs. vehicle on the same day after treatment; # p < 0.05, ## p < 0.01, vs. pretreatment (PRE) values.
Effect of Paclitaxel Treatment on Synaptic Transmission in the ACC
There were no statistically significant differences between the untreated group and the vehicle-treated group in the fEPSP initial slope (Emax), which was used as an indication of excitatory synaptic strength (1.48 ± 0.26 and 1.30 ± 0.09 mV/m, respectively; p > 0.05; results not shown). Therefore, untreated rats were used as the control group throughout the study. The excitatory synaptic strength from paclitaxel-treated rats was significantly stronger than that from untreated rats, as evidenced by a statistically higher Emax (2.45 ± 0.65 mV/ms) in paclitaxel-treated rats compared to untreated rats (1.48 ± 0.26 mV/ms; p < 0.001; fig. 2).
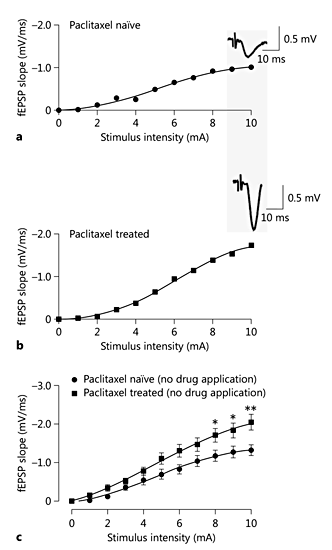
The effect of paclitaxel treatment on fast synaptic transmission in the ACC. a, b Representative stimulus-response curves from an ACC slice from an untreated rat (control) and a paclitaxel-treated rat. Inserts are sample fEPSPs taken at a stimulus intensity of 10 mA in the respective slices. c Superimposed averaged stimulus-response curves showing slices from paclitaxel-treated and untreated rats. Each point represents the mean ± SEM of the values obtained from 7-14 slices. * p < 0.05, ** p < 0.01, vs. control.
The effect of paclitaxel treatment on fast synaptic transmission in the ACC. a, b Representative stimulus-response curves from an ACC slice from an untreated rat (control) and a paclitaxel-treated rat. Inserts are sample fEPSPs taken at a stimulus intensity of 10 mA in the respective slices. c Superimposed averaged stimulus-response curves showing slices from paclitaxel-treated and untreated rats. Each point represents the mean ± SEM of the values obtained from 7-14 slices. * p < 0.05, ** p < 0.01, vs. control.
Effect of Paclitaxel Treatment on the GABAergic System in the ACC
Treatment with GABA (0.4 µM) significantly reduced the fEPSP slope of ACC slices from paclitaxel-treated rats (p < 0.001 vs. control; fig. 3b) and restored their excitability to a level similar to that in slices from untreated rats, as the value of Emax significantly decreased by 68.20 ± 1.63% (p < 0.001 vs. control; fig. 3b). However, this treatment did not cause any significant changes in the stimulus-response curve obtained from slices from untreated rats (change in Emax = 8.31 ± 0.23%; p > 0.05 vs. control; fig. 3a). Conversely, the bath application of the GABAB antagonist CGP 55845 (1 µM) caused a significant increase in the value of Emax in slices from paclitaxel-naïve rats by 71.15 ± 6.08% (p < 0.01 vs. control) but had no statistically significant effect in slices from paclitaxel-treated rats (change in Emax = -3.67 ± 2.29%; p < 0.05 vs. control; fig. 4).

The effect of bath application of GABA (0.4 µM) on the stimulus-response curves of fEPSPs in paclitaxel-naïve (a) and paclitaxel-treated rats (b). GABA significantly decreased the maximum response (Emax; ** p < 0.01) in slices from paclitaxel-treated rats but had no such effect in slices from untreated rats.
The effect of bath application of GABA (0.4 µM) on the stimulus-response curves of fEPSPs in paclitaxel-naïve (a) and paclitaxel-treated rats (b). GABA significantly decreased the maximum response (Emax; ** p < 0.01) in slices from paclitaxel-treated rats but had no such effect in slices from untreated rats.

The GABAB antagonist CGP 55845 (1 µM) caused a significant increase in excitability in ACC slices from paclitaxel-naïve rats, as shown by a significant increase in the maximum response (Emax; ** p < 0.01) (a) but had no significant effect in the same slices obtained from rats pretreated with paclitaxel (b).
The GABAB antagonist CGP 55845 (1 µM) caused a significant increase in excitability in ACC slices from paclitaxel-naïve rats, as shown by a significant increase in the maximum response (Emax; ** p < 0.01) (a) but had no significant effect in the same slices obtained from rats pretreated with paclitaxel (b).
Effect of E139 on Synaptic Transmission in the ACC
Consistent with our previous report [10], 10 min of bath perfusion of E139 (10 µM) caused a long-lasting depression in glutamate-mediated excitatory synaptic transmission (44.74 ± 9.1% decrease from baseline caused by bath application of 10 μM E139; p < 0.05; fig. 5a) as measured by the initial slope of the fEPSPs of ACC slices obtained from rats not treated with paclitaxel. This effect peaked 5-6 min after commencement of perfusion with E139 and lasted for over 50 min after the washout. This effect of E139 on fEPSP slopes was concentration dependent with an Emax value of about 47.3%. The calculated EC50 value was 2.25 µM (fig. 5b). Bath application of E139 (10 µM) to ACC slices obtained from paclitaxel-treated rats caused a depression in fEPSP slopes of 28.99 ± 6.1% (p < 0.05). Although this value is less than that obtained with untreated rats, the difference is not statistically significant (p > 0.05; fig. 5c).
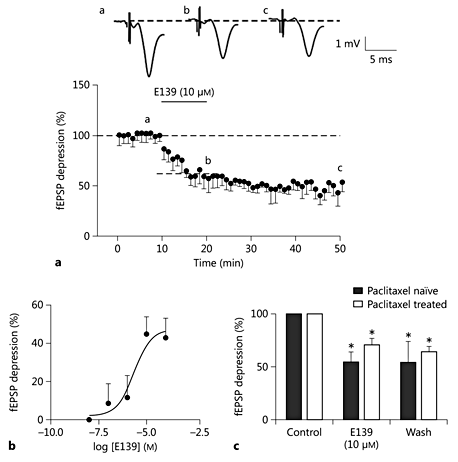
E139 caused a consistent depression in fEPSPs in the rat ACC. a Normalized and averaged time-effect plots from paclitaxel-naïve (n = 5) rats showing the effect of E139 (10 μM) on fast synaptic transmission. Inserts are sample fEPSPs (average of 5 consecutive traces) recorded in the area II/III of the rat ACC taken at the times indicated by letters in the graph. b Concentration-response curve of the effect of E139 on ACC slices from an untreated rat. Each point on the graph represents the mean of 3-5 slices. c Summary bar graph showing the effect of E139 on fEPSPs in paclitaxel-naïve and paclitaxel-treated rats. * p < 0.05, ** p < 0.01, vs. baseline.
E139 caused a consistent depression in fEPSPs in the rat ACC. a Normalized and averaged time-effect plots from paclitaxel-naïve (n = 5) rats showing the effect of E139 (10 μM) on fast synaptic transmission. Inserts are sample fEPSPs (average of 5 consecutive traces) recorded in the area II/III of the rat ACC taken at the times indicated by letters in the graph. b Concentration-response curve of the effect of E139 on ACC slices from an untreated rat. Each point on the graph represents the mean of 3-5 slices. c Summary bar graph showing the effect of E139 on fEPSPs in paclitaxel-naïve and paclitaxel-treated rats. * p < 0.05, ** p < 0.01, vs. baseline.
Discussion
In this study on paclitaxel-induced mechanical allodynia, there was increased excitability in the ACC, a brain region known to be involved in pain perception and modulation. This increased excitability was reversed by addition of exogenous GABA, returning the excitability to a level similar to control. The results also showed that the anilino enaminone (E139), which works partly by enhancing extracellular GABA, depressed fEPSPs in both control and paclitaxel-treated ACC, with less effect in the latter.
Using a regimen with a low dose of paclitaxel [8 mg/kg on 2 alternate days (cumulative dose of 16 mg/kg)] did not produce any mortality but still produced significant mechanical allodynia in rats. A dose of paclitaxel higher than that used in our study [16 mg/kg on 2 alternate days (cumulative dose of 32 mg/kg)] also produce mechanical allodynia in rats [12], but mortality rate was approximately 9% with this regimen. Hence, the cumulative dose of 16 mg/kg should be the preferred dosage for this experiment. However, this study showed that exposing rats to paclitaxel caused mechanical allodynia accompanied by significant electrophysiological changes in the ACC in rats in vitro. These changes were observed as an upward shift in the stimulus-response curve, which was evident as an increase in the relationship between the stimulus intensity and the maximum fEPSP slope. The increase in Emax along with the generally higher responses at lower stimulus intensities in slices from paclitaxel-treated rats reflected an increased excitability of the ACC in paclitaxel-treated rats. This finding is consistent with both imaging and neurophysiologic studies that had also shown increased activity in the ACC in patients and animal models of neuropathic pain [4,5].
GABA is the major inhibitory neurotransmitter in the CNS and has a regulatory effect on the level of neuronal excitability [14]; therefore, the observed increased excitability in the ACC of paclitaxel-treated rats may be due to disinhibition arising from a loss of the tonic GABA effect in the region. The addition of GABA, at a physiologically relevant concentration in the ACC [15], to slices from paclitaxel-treated rats restored the excitability of the ACC to a level similar to that in untreated rats, but had no effect on ACC slices from untreated animals. This indicates that there is a deficiency in GABA in the ACC in paclitaxel-treated rats. The deficiency in GABA in the ACC of paclitaxel-treated rats could have been due to the increased expression of GABA transporter 1 (GAT-1) in the ACC [6]. GAT-1 expression was also shown to increase in other CNS areas, such as the spinal cord of rats with PINP [16]. The deficiency in GABA in the ACC in paclitaxel-treated rats is further supported by the observation that the GABAB antagonist CGP 55845 increased the excitability level in untreated rats but not in paclitaxel-treated ones, where the GABAergic response had possibly been dysregulated by paclitaxel treatment. The increased excitability and altered GABA content of the ACC could be due to an increased nociceptive input from the peripheral nerves damaged by paclitaxel, rather than by direct CNS toxicity, since paclitaxel has limited ability to cross the blood-brain barrier [17] because the ABC (ATP-binding cassette) active transporter P-glycoprotein limits its entry in the CNS [18].
The enaminone E139 was found to attenuate PINP in rats [13], and it also enhanced extracellular GABA levels in vitro [10]. In the current study, E139 depressed fast synaptic transmission in the ACC of slices from rats treated with paclitaxel, but to a less extent compared to controls. This decreased activity of E139 may be attributed to increased GAT-1 activity in the ACC caused by paclitaxel treatment [6]. Although E139 is speculated to act as a GAT-1 inhibitor [10], paclitaxel may be enhancing GAT-1 expression and activity to an extent that E139 effects are partially occluded or blunted. It is also plausible that the observed (residual) effect of E139 in slices from paclitaxel-treated rats is via other mechanisms that this compound is known to have [9], and these additional actions may contribute to a better effect of E139 in PINP [13].
Conclusion
Paclitaxel-treated rats developed mechanical allodynia and exhibited increased excitability in the ACC region. This increase in excitability appeared to be due to a deficiency in GABA-mediated neurotransmission in the ACC, thus indicating that paclitaxel could cause supraspinal changes in synaptic transmission via modulation of GABAergic mechanisms involving GABAB receptors. These changes probably contributed to the pathophysiology of PINP. Further research to evaluate both the GABAergic system as a potential therapeutic target and E139 as a therapeutic drug for managing PINP is warranted.

