

Abstract
Background: Valproic acid (VPA), an established antiepileptic drug, was assessed for antitumor activity, including its effects on glioblastoma, but its role has not been determined. Methods: In the present study, we investigated VPA-induced apoptosis effects on human U87 cells by cell viability, lactate dehydrogenase (LDH) release, TUNEL/Hoechst staining and flow cytometric in vitro, then we further explored the underlying molecular mechanisms using the selective antagonists PD98059, LY294002 and SB216763. Results: The data showed that VPA dose-dependent induction of glioma U87 cells to undergo apoptosis through the mitochondria-dependent pathway in vitro. VPA activated the ERK/Akt pathways by increasing their protein phosphorylation and in turn inhibited GKS3β activation by the induction of GKS3β phosphorylation. However, the MAPK inhibitor PD98059 and/or PI3K inhibitor LY294002 were able to antagonize the effects of VPA by abolishing ERK/Akt activations and cancelling GSK3β suppression, thus it impaired VPA apoptosis-inducing effects on glioma cells. Furthermore, the GSK3β inhibitor SB216763 caused a strong suppression of GSK3β activity, which showed similar effects of VPA on regulation of protein expression and apoptosis. Conclusion: These findings suggest that GSK3β may be the central hub for VPA-induced apoptosis and VPA can be further evaluated as a novel agent for glioma therapy.
Introduction
Glioma is the most common and deadly primary malignancy in the central nervous system (CNS) [1]. Clinically, glioma, particularly the aggressive form of high-grade glioma (glioblastoma, GBM), has an extremely poor prognosis [2]. Over the past few decades, multimodality treatment, using combination of extensive tumor mass resection, radiation therapy, and chemotherapy, has shown little improvement in well being and led median patient survival to only under 15 months [3,4]. Even the most frequently used first-line treatment drug, Temozolomide (TMZ) for GBM may only increase an average of two month survival of patients [4]. Thus, it is urgently needed to identify novel strategies to treatment glioma.
Valproic acid (VPA) is a clinically established drug for the treatment of epileptic seizure [5,6] and epileptic seizure could occur in approximately 50% of GBM patients [7,8]. One of the mechanisms of VPA antiepileptic activity is known to inhibit activities of histone deacetylase (HDAC) and DNA methyltransferase [9]. Interestingly, increasing evidence suggests that VPA had an anticancer effect in several GBM cell lines [10]. VPA treatment induced cell cycle arrest and enhanced the efficiency of glioma radiotherapy in clinical trials [11]. Also, VPA may show antineoplastic activity based on its gene-regulation functions [12,13,14,15,16]. Expression of CD44 and CD56 [17], p21 and topoisomerase-II [18] could be regulated after VPA administration in GBM cell lines. Treatment with VPA in human neuroblastoma cells and human leukemic cell lines resulted in a strong inhibition of cell proliferation and induction of cell differentiation and apoptosis [19,20,21]. In other human cancer cell lines, administration of VPA inhibited cancer cell growth [22,23], induced apoptosis-regulatory and differentiation gene expression [24] and decreased angiogenic potency [25].
It was shown that VPA anti-tumor effects may be achieved by stimulating multiple death pathways including both caspase-dependent and -independent apoptotic signaling pathways [21]. However, in GBM, it is still unclear how does VPA initiate the apoptosis process, key signaling pathway (s) and regulation factor (s) are still uncharted. Thus, the present study was designed to assess the effects of VPA on inducing apoptosis using one of the most common human glioma cell lines, U87. Furthermore, we investigated if extracellular signal-regulated kinases (ERK), protein kinase B (Akt) and glycogen synthase kinase-3β (GSK3β) are involved in the mechanism of activity of VPA.
Materials and Methods
Cell line and culture
The human glioma U87-MG cell line was obtained from the American Type Culture Collection (Rockville, MD) and cultured in Dulbecco's modified Eagle's medium (DMEM supplemented with 10% fetal bovine serum (FBS), 100 U penicillin, and 100 U streptomycin (Sigma-Aldrich Chemical Co., St Louis, MO) at 37°C in a humidified atmosphere, with 5% CO2 and 95% air.
Cell viability assay
Cells were seeded and grown overnight and treated with 2-16 mM VPA (Sigma-Aldrich Chemical Co., St Louis, MO), 50 μM of TMZ (Sigma-Aldrich Chemical Co.), or negative control (solvent only) for up to 72 h. At the end of each experiment, cell viability was assessed using the cell viability assay kit (Promega, Madison, WI), according to the manufacturer's instructions. Briefly, 25 µL of treated cell samples were collected to which was added 5 µL of cell viability assay reagent per well. The plates were incubated at 37°C to allow the cells to convert. Then the fluorescent signal was measured with an automatic microplate reader (BioTek Instruments, Winooski, VT). The data were summarized and expressed as percentage of the control.
Lactate dehydrogenase (LDH) release assay
VPA cytotoxicity was determined by the level of lactate dehydrogenase (LDH), the stable cytosolic enzyme released upon cell lysis. LDH was quantitatively measured with the use of a cytotoxicity assay kit (Promega) according to the manufacturer's instructions. 50 μL supernatant of treated cell samples was transferred to a 96-well plate. An equal volume of cytotoxicity assay reagent was added to each well and incubated for 30 min at 37°C, and the reaction was stopped by adding stop buffer from the kit. The absorbance rate was measured at 490 nm using a plate reader (BioTek Instruments). A maximum LDH release control was set by adding 10 × lysis buffer to lyse all cells 45 min before the procedure. The data are expressed as a percentage of the maximum LDH release value.
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay
The level of VPA-induced apoptosis in glioma cells was assessed using an in situ cell death detection kit (Roche, Mannheim, Germany), according to the manufacturer's instructions. In brief, U87 cells were grown on coverslips and treated with VPA or controls for up to 72 h. The cells were then fixed in freshly made 4% paraformaldehyde in phosphate-buffered saline (PBS, pH 7.4) with gentle shaking for 60 min at room temperature and rinsed with PBS and treated with 0.1% Triton X-100 in 0.1% sodium citrate on ice for 2 min. Next, the cells were incubated with a fluorescein TUNEL-TdT reaction mixture in a humidified chamber at 37°C for 60 min and then reviewed and scored under a Leica fluorescence microscope (Buffalo Grove, IL). The number of TUNEL-positive cells was counted in six different fields. The experiments were repeated six times and the data were summarized as % of control.
Hoechst staining
Cell apoptosis was further characterized with Hoechst 33342 (Molecular Probes, USA) staining of nuclear chromatin. Cells were grown on coverslips and treated with VPA or controls for up to 72 h and then fixed with 4% formaldehyde for 10 min. After being washed with PBS 3 times, the cells were stained with Hoechst 33342 (1 μg/mL in PBS and 0.1% Triton X-100) for 15 min at room temperature and intact, fragmented, or condensed DNA was counted under a Leica fluorescence microscope. The experiments were repeated six times. The data are expressed as mean ± S.E. and reported as % of control.
Apoptosis flow cytometry assay
An Annexin V-fluorescein isothiocyanate/propidium iodide (FITC/PI) apoptosis detection kit was obtained from Sigma-Aldrich and used to detect apoptotic cells. Briefly, cells were harvested 72 h after VPA treatment, centrifuged, washed twice with PBS, and then resuspended in Annexin V-binding buffer. FITC-conjugated Annexin V and PI were added to the cells, incubated for 30 min at room temperature in the dark and analyzed with a flow cytometer (BD Biosciences, Franklin Lakes, NJ). The experiments were repeated at least 3 times and data were summarized as % of control.
Protein extraction and Western blot
Total cell lysis was prepared using a lysis buffer (Cell Signaling Technology Inc., Beverly, MA), mitochondria was extracted using a mitochondria isolation kit (Thermo Fisher Scientific Inc., Waltham, MA). The protein concentration was measured using a BCA protein assay kit (Thermo Fisher Scientific Inc.). An equal amount of protein (60 μg/sample) was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) in 10% SDS-PAGE gels and transferred to polyvinylidene difluoride (PVDF) membranes (Thermo Fisher Scientific Inc.). The membranes were then incubated in 5% nonfat dry milk in Tris-buffered saline with 0.1% Triton X-100 (TBST) for 1 h at room temperature and incubated with primary antibodies dilutions in TBST at 4°C overnight. The primary antibodies were anti-ERK1/2 (1:1000), phospho-ERK1/2 (1:500), Akt (1:1000), phospho-Akt (1:1000; all from Cell Signaling Technology Inc.), GSK3β (1:500), phospho-GSK3β (pSer9) (1:500; all from Sigma-Aldrich Chemical Co.), caspase-3 (1:1000), cleaved-caspase-3 (1:500), caspase-9 (1:1000), cleaved-caspase-9 (1:500), Bax (1:500), Bcl-2 (1:500), cytochrome-c (1:500), AIF (1:1000), cleaved-PARP (1:1000), and β-actin (1:2500; all from Cell Signaling Technology Inc.). After being washed in TBS-T thrice, the membranes were further incubated with a secondary antibody (Cell Signaling Technology Inc.) at room temperature for 1 h. To visualize the protein bands, membranes were briefly incubated with Super Signal West Pico Chemiluminescence Substrate (Thermo Fisher Scientific Inc.) and quantified using Image J software (National Institute of Heath, Bethesda, MD).
Statistical analysis
All statistical analyses were performed using SPSS 18.0, a statistical software package from SPSS, Inc. (Chicago, IL). The data were presented as mean ± SD and statistically analyzed using one-way ANOVA followed by the Student-Newman-Keuls test. P<0.05 was considered statistically significant.
Results
VPA dose-dependent induction of glioma U87 cells to undergo apoptosis in vitro
In this study, we first confirmed the effects of VPA on regulation of glioma cell apoptosis in vitro by treating U87 cells with VPA (2-16 mM) or a positive control (TMZ at 50 μM) [10,26]. We found that after 72 h of treatment, VPA markedly reduced tumor cell viability but increased the LDH release in a concentration-dependent manner (P < 0.05) (Fig. 1A and 1B). However, the 72 h VPA treatment showed significant quantities of obvious TUNEL-positive cells (Fig. 1C and 1E) or Hoechst-positive cells (Fig. 1D and 1F) compared to the control cells (P < 0.05). In addition, compared to 50 μM TMZ positive control, 4 mM VPA treatment was able to achieve similar effects of TMZ on cell viability, LDH release, and TUNEL and Hoechst staining.

Effects of VPA treatment on induction of glioma cell apoptosis. U87 cells were cultured and treated with VPA (2-16 mM) or a positive control TMZ (50 μM). After 72 h, cell viability (A) and LDH release assays (B) were measured. TUNEL staining (C and E) was performed to detect and quantify apoptosis cells. Morphology changes were evident by Hoechst 33342 staining, and the arrow indicates karyopycnosis and nuclear fragmentation (D and F). Scale bar: 25 μM. The data are presented as means ± SD of six experiments. *P<0.05 vs. control group.
Effects of VPA treatment on induction of glioma cell apoptosis. U87 cells were cultured and treated with VPA (2-16 mM) or a positive control TMZ (50 μM). After 72 h, cell viability (A) and LDH release assays (B) were measured. TUNEL staining (C and E) was performed to detect and quantify apoptosis cells. Morphology changes were evident by Hoechst 33342 staining, and the arrow indicates karyopycnosis and nuclear fragmentation (D and F). Scale bar: 25 μM. The data are presented as means ± SD of six experiments. *P<0.05 vs. control group.
Flow cytometric Annexin V-FITC/PI data showed no statistical difference among the numbers of necrotic cells in VPA and TMZ treatment groups (data not shown, P > 0.05) (Fig. 2A; Annexin V-FITC-/PI+ cells in Quadrant 1 were necrosis), whereas VPA-treated U87 cells exhibited significantly higher apoptosis in a dose-dependent manner, compared to controls (P < 0.05) (Fig. 2B). Treatment with 4 mM VPA resulted in approximately 25% of U87 cells exhibiting apoptosis, a result similar to the TMZ group and identical to the results of TUNEL and Hoechst staining in Figure 1. Thus, we selected this dose for the following in vitro experiments.

Flow cytometric Annexin V-FITC/propidium iodide (PI) double staining detection of glioma cell apoptosis after VPA treatment. Annexin V-FITC-/PI- (Quadrant 4) cells were alive, and Annexin V-FITC+/PI- cells were considered to be in the early stages of apoptosis (Quadrant 3), while Annexin V-FITC+/ PI+ cells (Quadrant 2) were in the late stages of apoptosis. Annexin V-FITC-/ PI+ cells (Quadrant 1) were necrotic. The total apoptosis rate was calculated as Quadrant 2 + Quadrant 3. The data are presented as means ± SD of six experiments. *P<0.05 vs. control group.
Flow cytometric Annexin V-FITC/propidium iodide (PI) double staining detection of glioma cell apoptosis after VPA treatment. Annexin V-FITC-/PI- (Quadrant 4) cells were alive, and Annexin V-FITC+/PI- cells were considered to be in the early stages of apoptosis (Quadrant 3), while Annexin V-FITC+/ PI+ cells (Quadrant 2) were in the late stages of apoptosis. Annexin V-FITC-/ PI+ cells (Quadrant 1) were necrotic. The total apoptosis rate was calculated as Quadrant 2 + Quadrant 3. The data are presented as means ± SD of six experiments. *P<0.05 vs. control group.
VPA activation of ERK and Akt but inhibition of GSK3β in glioma cells
Then we explored VPA gene regulation in glioma cells and found that multiple signal transduction pathways were involved in VPA-mediated apoptosis in U87 cells (Fig. 3A) and that, after 72 h of VPA treatment, ERK and Akt proteins were phosphorylated, indicating that their activities were induced (Fig. 3B and 4C) but GSK3β activity was inhibited, because of increased GSK3β phosphorylation (Fig. 3D) (all P < 0.05). To further determine whether GSK3β suppression was due to ERK/Akt activation, simultaneously, we co-treated U87 cells with VPA plus a MAPK inhibitor PD98059 and/or a PI3K inhibitor LY294002 (both at 50 μM, according to a previous study [27]). After 72 h of treatment, PD98059 or LY294002 markedly attenuated the level of VPA-induced ERK and Akt phosphorylation and impaired GSK3β phosphorylation (all P < 0.05). As shown in Figure 3D, treatment with the combination of the two inhibitors completely abolished VPA-induced GSK3β inhibition by phosphorylation (P < 0.05).
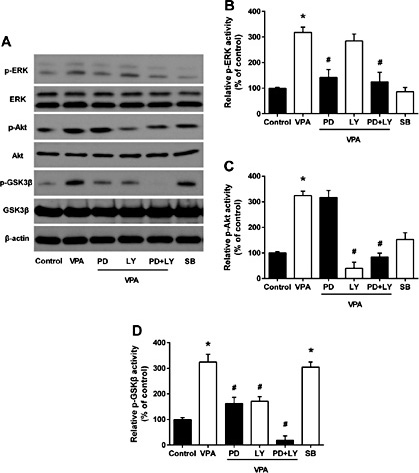
Effects of VPA treatment on modulation of protein expression. The data showed multiple signal transduction pathways modified by VPA and inhibitors. Western blot analysis (A). Quantification of Western blot data for p-ERK activity (B), p-Akt activity (C), p-GSK3β activity (D). The data are presented as means ± SD of six experiments. *P<0.05 vs. control group; #P<0.05 vs. VPA group. PD, PD98059; LY, LY294002; SB, SB216763.
Effects of VPA treatment on modulation of protein expression. The data showed multiple signal transduction pathways modified by VPA and inhibitors. Western blot analysis (A). Quantification of Western blot data for p-ERK activity (B), p-Akt activity (C), p-GSK3β activity (D). The data are presented as means ± SD of six experiments. *P<0.05 vs. control group; #P<0.05 vs. VPA group. PD, PD98059; LY, LY294002; SB, SB216763.

Effects of VPA on activation of intrinsic mitochondrial apoptosis pathway proteins. Western blot (A). Quantification of Western blot data for cleaved caspase-3 and cleaved capase-9 (B and C), anti- and pro-apoptosis protein Bax and Bcl-2 (D and E), mito-CytC (mitochondria cytochrome c) and cyto-CytC (cytosol cytochrome c) (F and G), and AIF and cleaved PARP (H and I). The data are presented as means ± SD of six experiments. *P<0.05 vs. control group; #P<0.05 vs. VPA group.
Effects of VPA on activation of intrinsic mitochondrial apoptosis pathway proteins. Western blot (A). Quantification of Western blot data for cleaved caspase-3 and cleaved capase-9 (B and C), anti- and pro-apoptosis protein Bax and Bcl-2 (D and E), mito-CytC (mitochondria cytochrome c) and cyto-CytC (cytosol cytochrome c) (F and G), and AIF and cleaved PARP (H and I). The data are presented as means ± SD of six experiments. *P<0.05 vs. control group; #P<0.05 vs. VPA group.
VPA activation of the mitochondria apoptosis in glioma cells in vitro, ERK/Akt pathways and GSK3β contributed to VPA-induced apoptosis
Next, we assessed the mechanism of VPA-induced apoptosis in glioma cells by measured apoptosis-related protein levels (Fig. 4A). We found that VPA induced expression of cleaved caspase 3 and cleaved caspase 9 in U87 cells (Fig. 4B and 4C). Moreover, the expression of Bcl-2 family protein Bax was increased, whereas Bcl-2, the anti-apoptotic member, was decreased by VPA treatment (Fig. 4D and 4E). The release of cytochrome c from the mitochondria after VPA treatment has also been increased (Fig. 4F and 4G). The expression of apoptosis-inducing factor (AIF) and ribose polymerase (PARP) was also up-regulated (Fig. 4H and 4I) (all P < 0.05). These data suggested that VPA was able to activate the mitochondria-mediated apoptosis pathway.
Further, we assessed the expression of apoptosis-related proteins after the combination of a MAPK inhibitor PD98059, a PI3K inhibitor LY294002, or a GSK3β inhibitor SB216763 with VPA treatment. MAPK inhibitor together with PI3K inhibitor was able to alleviate VPA-induced apoptosis, with a decrease in cleaved caspase-3, cleaved caspase-9, anti-apoptosis protein Bax, cytosol cytochrome c, AIF, cleaved PARP, but with up-regulation of pro-apoptosis protein Bcl-2 and mitochondria cytochrome c (all P < 0.05). Meanwhile, GSK3β inhibitor was able to regulate expression of apoptosis factors (all P < 0.05), which has a similar effect to VPA treatment.
Inhibitors of MAPK and PI3K pathways abolished VPA induced apoptosis effects, but GSK3β inhibitor mimicked effects of VPA
MAPK inhibitor PD98059 with PI3K inhibitor LY294002 treatment partially or almost completely neutralized the VPA apoptosis-inducing effects (Fig. 5). However, direct treatment of U87 cells with a GSK3β inhibitor SB216763 20 μM strongly affect tumor cell apoptosis as shown in cell viability (Fig. 5A), LDH release (Fig. 5D), TUNEL/Hoechst staining (Fig. 5C and flow cytometry (Fig. 5E) (all P < 0.05).

Modulation of glioma cell apoptosis using different inhibitors with and without VPA. The MAPK inhibitor and PI3K inhibitor together with VPA or GSK3β inhibitor were administered to glioma cells and then subjected to cell viability (A), LDH release (B), TUNEL staining (C), Hoechst staining (D), and flow cytometry (E). *P<0.05 vs. control group; #P<0.05 vs. VPA group.
Modulation of glioma cell apoptosis using different inhibitors with and without VPA. The MAPK inhibitor and PI3K inhibitor together with VPA or GSK3β inhibitor were administered to glioma cells and then subjected to cell viability (A), LDH release (B), TUNEL staining (C), Hoechst staining (D), and flow cytometry (E). *P<0.05 vs. control group; #P<0.05 vs. VPA group.
Discussion
Apoptosis is an exquisitely programmed and orchestrated cellular process that creates a fragile balance between cell division and cell death to maintain homeostasis in the human body. Apoptosis is also a crucial event in many diseases. For example, increased levels of apoptosis induce degenerative diseases, such as Alzheimer's disease or Parkinson's disease, whereas down-regulated levels of apoptosis can contribute to tumorigenesis [29]. In GBM, apoptosis is inhibited or attenuated by several mechanisms of action, because cell survival promoting molecular chaperones is overexpressed [30,31,32]. Thus, to effectively control GBM, one possible strategy is to recreate harmony by induction or restoration of the apoptosis-signaling pathways to induce GBM cell apoptosis. However, up-to-date management of GBM did not achieve this goal [33]. Radical/extended surgical resection of tumor lesion followed by radiotherapy and concomitant and adjuvant chemotherapy with TMZ is a standard regimen for the treatment of GBM, but did not achieve the desired effects on the control of GBM in a clinical setting [34,35,36]. TMZ, an alkylating agent that is a first-line treatment option for GBM, has been available in the US for over two decades and shows an ability to induce DNA methylation damage and trigger tumor cell death through autophagy, apoptosis, and necrosis [37,38]. Clinically, TMZ showed a certain level of benefits as adjuvant radiotherapy in the improvement of 5-year survival rates of glioma patients [36], but this may not be true of glioma in children [39]. In our current study, we assessed the effects of VPA on glioma cells in vitro. VPA is a simple branched-chain fatty acid with a well-established efficacy for seizures. It has been proven to be one of the safest and best-tolerated drugs to date for epilepsy treatment, because VPA can easily and extensively penetrate the blood-brain barrier [40]. Clinically, VPA is widely used to control seizures provoked by brain tumors, including glioma, and surprisingly, it has been shown to have potential radiotherapy benefit in TMZ-resistant malignant glioma [10,11,41]. VPA had an anti-cancer effect in U87 at low dosages of the drug, U87 cells may be more sensitive to VPA compared to other GBM lines [10]. Thus, we applied U87 cells models to validate that VPA was able to reduce glioma cell viability and for the first time, we demonstrated that VPA activated ERK/Akt proteins and in turn inhibited GKS3β activity to induce glioma cells to undergo apoptosis.
There are two main apoptosis pathways in cells, i.e., the intrinsic mitochondrial pathway or external cell membrane pathway [10,11]. In the mitochondrial apoptosis pathway, the main characteristic is the increased mitochondrial permeability, release of cytochrome c, and pro- apoptotic/anti-apoptotic molecule modification. The balance between pro-apoptotic proteins, like Bax, Bak, and Bad, and anti-apoptotic proteins, including Bcl-2, Bcl-X, and Bcl-W are crucial for the regulation of cell survival and death [42]. Through the induction and formation of the apoptosis complex known as apoptosome, these apoptosis factors will activate caspase 9 and 3 to degrade genomic DNA and commit cell suicide [32,43,44]. We found that VPA treatment of glioma cells in vitro activated the intrinsic mitochondria apoptosis pathway, and blocked expression of pro-survival proteins (Bcl-2 and Bcl-x), but increased expression of the pro-apoptotic protein Bax.
Furthermore, the puzzle is how VPA initiates intrinsic mitochondria apoptosis-related proteins and which signal transduction pathway(s) contribute(s) to this process. Previous studies showed that VPA could activate multiple signal transduction pathways, including HDACs, glycogen synthase kinase 3 (GSK3α and β), protein kinase B (Akt), extracellular signal-regulated kinases (ERK), γ-aminobutyric acid, and oxidative phosphorylation [45,46,47,48,49,50,51,52,53]. These pathways can affect critical cellular events, such as cell growth, differentiation, apoptosis, and immunogenicity [54,55,56,57]. In our current study, we found that VPA treatment of glioma cells in vitro activated ERK and Akt proteins, but inhibited GSK3β activity. Thus, further investigation of the crosstalk among these cell signal transduction pathways is essential for elucidating the mechanisms of VPA-induced apoptosis. Indeed, the MAPK/ERK pathway is one of the best-studied pathways, which links to very fundamental cell processes, i.e., cell growth, differentiation, migration, and apoptosis [58,59,60,61]. Similarly, GSK3β functions in several cell events via self-phosphorylation, and in the brain, it works as a sensor to determine cell fate [62]. The activation of the MAPK pathway by different stimuli may lead to GSK3β inhibition [58,59,63]. In addition, as the major target of the PI3K, Akt regulates many downstream targets for the execution of its cell functions (such as cell growth and migration) [64,65,66]. Previous studies showed that Akt activation led to GSK3β phosphorylation [67,68], while others showed that lithium was able to activate PI3K and Akt and in turn to induce GSK3β phosphorylation and inactivation [69,70]. Our current data confirmed that the VPA treatment of glioma cells in vitro activated ERK and Akt proteins but inhibited GSK3β activity. Either MAPK or PI3K inhibitor was able to independently and partly restore GSK3β activity in VPA-treated glioma cells, while the ERK and Akt pathways both contributed to GSK3β suppression. For example, using the MAPK and PI3K inhibitors together, ERK/Akt activation and GSK3β suppression induced by VPA were cancelled. Simultaneously, the apoptosis effect initiated by VPA faded away, and there was no mitochondrial cytochrome c release that occurred in glioma cells, leading to absence of the mitochondria apoptosis pathway activation. Our current data are consistent between GSK3β suppression and apoptosis level, indicating that GSK3β is closely associated with the mitochondria-initialed apoptosis pathway and that it played a central role in mediating VPA-induced apoptosis in glioma cells (Fig. 6).
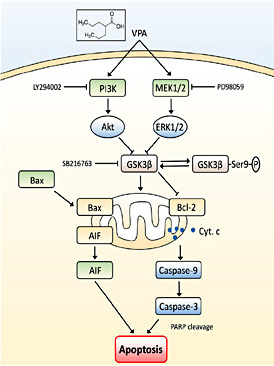
Illustration of GSK3β as a central apoptosis process for VPA treatment. VPA was able to activate the ERK and Akt pathways, and then lead to GSK3β suppression (phosphorylation) eventually, initiate the mitochondrial apoptosis process. MAPK inhibitor PD98059 and PI3K inhibitor LY294002 were able to antagonize VPA-induced GSK3β suppression and abolish VPA-induced apoptosis in glioma cells. However, GSK3β inhibitor SB216763 was able to directly suppress GSK3β activity and mimic effects of VPA on induction of glioma cell apoptosis.
Illustration of GSK3β as a central apoptosis process for VPA treatment. VPA was able to activate the ERK and Akt pathways, and then lead to GSK3β suppression (phosphorylation) eventually, initiate the mitochondrial apoptosis process. MAPK inhibitor PD98059 and PI3K inhibitor LY294002 were able to antagonize VPA-induced GSK3β suppression and abolish VPA-induced apoptosis in glioma cells. However, GSK3β inhibitor SB216763 was able to directly suppress GSK3β activity and mimic effects of VPA on induction of glioma cell apoptosis.
One of our previous studies showed that VPA markedly up-regulated ERK and Akt activity and protected neurons from trauma-induced brain injury in adult rat cerebral cortex [71]. The branch point between VPA-induced protection of normal neurons and VPA-induced glioma cell apoptosis may be the differences in GSK3β expression. In different human cancers, including glioma, GSK3β has been found to consistently express and regulate cell differentiation or growth arrest [72]. Our data demonstrates that either direct inhibition of GSK3β, using an inhibitor, or indirect inhibition, using VPA, through activation of the ERK/Akt pathways was able to trigger mitochondria apoptosis activation and induce U87 cells apoptosis. Although GSK3β-specific inhibitor SB216763 showed promising apoptosis-inducing effects in glioma cells in vitro, the mechanisms have not been fully elucidated. There is still a long way to go to apply it in clinical practice. VPA, which has already been widely used in clinical settings, may be helpful to this dilemma. But, also, further study e.g. xenograft experiments in vivo and clinical testing are indispensable.
Conclusions
In our current study, we gathered evidence showing that VPA was able to induce apoptosis in glioma cells in a dose-dependent manner through the activation of the mitochondria apoptosis pathway. VPA can induce activation of the ERK and Akt pathways and GSK3β inhibition. Furthermore, modification of GSK3β by the ERK/Akt inhibitor or the GSK3β inhibitor can results in apoptosis. These data suggest that GSK3β may be the central hub for VPA-induced apoptosis and VPA can be further evaluated as a novel agent for glioma therapy. However, our current study is just proof of principle and has limitations, such as the cell line in vitro model. Comprehensive studies including other glioma cell lines or in vivo are still needed.
Acknowledgments
This work was supported in part by a grant from the The National Key Technology Research and Development Program of the Ministry of Science and Technology of China (2014BAI04B02).
Disclosure Statement
The authors declare that there is no conflict of interest in this work.

