

Abstract
Acute kidney injury (AKI) is a major cause of morbidity and mortality in hospitalized patients. Despite substantial progress being made in understanding the mechanisms contributing to the pathophysiology of AKI, we have so far been unsuccessful in devising adequate therapeutic strategies against the disease. A growing body of evidence suggests that the activation of mineralocorticoid receptors (MRs) may contribute to the exacerbation of AKI. Indeed, several studies have demonstrated the potential of MR antagonists in preventing and treating certain forms of experimental AKI. However, the main drawback of these medications is their side-effect profile. This has been addressed with the development of newer nonsteroidal MR antagonists, which have a comparable therapeutic profile without the side effects. This mini review aims at providing a brief overview of the rationale, potential benefits and challenges associated with the use of MR antagonists, particularly the novel nonsteroidal MR blockers, as therapy against AKI.
Introduction
Acute kidney injury (AKI) is responsible for up to 20% of all hospitalizations and is seen in more than 30% of critically ill patients worldwide [1]. While it was previously thought to be a self-limiting intra-renal phenomenon, it is now recognized to affect other vital organs, resulting in a high degree of systemic morbidity and mortality in the severely ill [2]. Moreover, even when one survives an episode of AKI, it results in an increased risk of long-term sequelae including the development of chronic kidney disease (CKD) and eventually end-stage renal disease and premature death [3]. Consequently, an ideal therapeutic strategy would attack all aspects of AKI; that is, prevent its development or limit its severity, promote renal recovery after the injury, and reduce its chronic sequelae. A key mechanism that appears to contribute to all of these facets of AKI is the persistent alteration in intrarenal perfusion that is driven by abnormal vasoreactivity of the renal microcirculation.
The Imbalance between Vasoconstrictors and Vasodilators in AKI - Role of Aldosterone
AKI is often instigated when renal perfusion falls to a level that results in tissue ischemia, as seen during organ transplantation, surgeries (particularly cardiac or vascular), and shock. The ischemic insult activates signaling cascades causing the release of pro-inflammatory cytokines, reactive oxygen species (ROS), chemokines, and leukocyte activation, which result in damage to the endothelium with ensuing microvascular dysfunction characterized by enhanced vasoconstriction and diminished vasodilation [4]. This imbalance between vasoconstriction and vasodilation further exacerbates renal hypoperfusion, particularly in the outer medullary region, extending the renal injury [4]. Moreover, it also facilitates sustained or repetitive bouts of hypoperfusion, thereby increasing the risk of chronic renal injury [5]. Hence, measures that can either re-establish normal vasodilation or reduce vasoconstriction may represent an effective therapeutic strategy against ischemia-reperfusion-induced AKI (I/R-AKI). Yet, despite several successes against experimental AKI, we have thus far not been effective in translating these strategies to clinical AKI. Recent advances in our understanding of the role played by aldosterone in I/R-AKI have opened up new therapeutic prospects to combat this disease.
Aldosterone is primarily known for its role in maintaining sodium and potassium homeostasis via its effects on the mineralocorticoid receptor (MR). While its actions were previously thought to be mainly limited to the distal tubules, it is now recognized to act on a variety of nonepithelial tissues including the vasculature [6]. For instance, it has been found to produce a dose-dependent contraction of vascular smooth muscle cells (VSMC) in humans via the MR receptor (hence spironolactone (SP) was able to block these effects) [7]. These vascular effects are also present in the kidney; indeed, Arima et al. [8] reported that aldosterone caused vasoconstriction of isolated-perfused afferent and efferent arterioles (although this appeared to be a nongenomic effect and thus may be independent of the MR). Aldosterone, also directly triggers pro-inflammatory and pro-fibrotic pathways, which foster the progression and transformation of acute to chronic injury [6]. Consequently, in conditions with high aldosterone levels, such as ischemic AKI, the excessive aldosterone may be promoting regional hypoperfusion, inflammation, and injury, thereby contributing to the various facets of the AKI spectrum [9]. Thus, the availability of aldosterone antagonists provides us with a potential therapeutic strategy against various diseases characterized by excessive aldosterone and hypoperfusion. Indeed, this prospect seems all the more enticing in that these same mechanisms also appear to be present in humans, as demonstrated by clinical studies that have found MR antagonists to exert some beneficial actions on the heart and kidney [10,11].
MR Antagonism in Experimental AKI
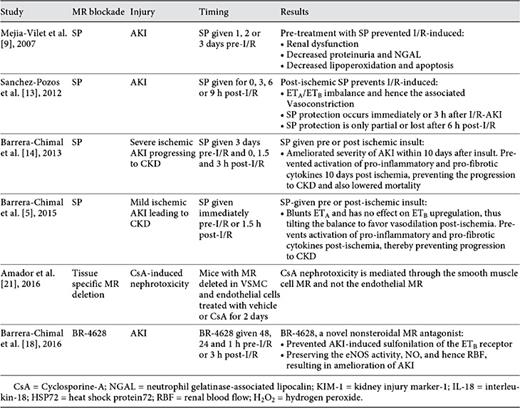
The concept that MR blockade may have the potential as AKI therapy was first explored by Mejia-Vilet et al. [9]. They tested whether blocking the MR receptor by pre-treating rats with SP ameliorated I/R-AKI. They found that the SP-treated animals exhibited less renal injury post I/R-AKI, as evidenced by decreased elevation of creatinine, proteinuria, urinary excretion of N-acetyl-β-D-glucosaminidase, apoptosis, and morphological injury. In a separate study, the same group found that the protective actions of SP can be replicated by performing adrenalectomy [12]; removing the adrenals suppressed baseline aldosterone levels and prevented ischemia-induced increases in aldosterone secretion, attenuating the renal injury, similar to MR antagonism. Taken together, these studies suggest that adrenal steroid production is responsible for MR activation during I/R-AKI, which in turn exacerbates renal injury. However, they did not establish whether MR blockade is effective when given after inducing I/R-AKI (thus making it more clinically relevant) and whether it would protect against the chronic sequelae of AKI, including progression to CKD. Sanchez-Pozos et al. [13] addressed the first question by administering SP at 0, 3, 6, and 9 h post I/R. They found that MR antagonism was effective at all time points, albeit the efficacy may have been decreasing at the later time points, raising the possibility that the protection may be time-dependent. The second query was answered by Barrera-Chimal et al. [14], who showed that MR blockade was effective at preventing the transformation of I/R-AKI to CKD.
Limitations of Available MR Antagonists and Alternative Approaches
While the above experiments suggest that MR blockade may be an appealing target to treat AKI, there are significant limitations that curb their use. The dose of SP used in animals (20 mg/kg body weight) is much higher than that used in humans, which raises the question of whether it will be clinically effective without compromising safety. Indeed, high doses of SP increases the risk of renal side effects (hyperkalemia and renal dysfunction) and also of its anti-progesterone and anti-androgen-dependent side effects (due to lack of selectivity toward other members of the oxosteroid family) [15]. Using eplerenone, a more selective MR antagonist, may confer less side effects, but may not be as effective since it is only 50-70% as potent as SP [16]. Moreover, MR antagonists increase aldosterone levels, which may exert nongenomic effects, as well as compete with the antagonist for binding to the MR and potentially decrease their utility.
These shortcomings of the steroidal MR antagonists have led to the search for alternative approaches of blocking MR activity, which may have fewer side effects (table 1). One approach has been to develop novel nonsteroidal MR antagonists. High throughput screening analyses have identified various dihydropyridine and pyrazoline derivatives that inhibit the MR in a highly selective and potent manner (up to 60 times more selective and 10 times more potent than SP). In addition, they have thus far been reported to have less side effects in preliminary clinical studies. One such compound, BR-4628, which was first optimized by Fagart et al. [17] from the dihydropyridine structure, has similar potency and minimal side effects to steroidal MR antagonists. Barrera-Chimal et al. [18], tested whether this drug would also protect against experimental I/R-AKI. They found it to be equally effective at preventing I/R-AKI (given 48 h before ischemia) or treating I/R-AKI (given 3 h after induction of ischemia). Moreover, they found it to have minimal side effects, including hyperkalemia (like the chronic studies [19]), increasing the possibility that they may be safely used clinically. Table 2 enumerates these aforementioned studies.

Potential Vascular Mechanisms by Which MR Antagonists Protect against AKI
As mentioned before, I/R-AKI is associated with a persistent decrease in renal perfusion that contributes to the renal injury in I/R-AKI. The improvement in AKI achieved with MR antagonism or adrenalectomy is associated with improved recovery of renal perfusion. One important mechanism by which AKI causes persistent renal hypoperfusion has been elucidated by the combined studies of Bobadilla's and Jaisser's labs. Their earlier studies demonstrated that MR activation is associated with significant changes in the activity of 2 important vasoactive systems; the nitric oxide (NO) and endothelin (ET) systems. They first provided evidence that NO is decreased during I/R-AKI by a MR-dependent mechanism; I/R-AKI-induced decreases in urinary NO2/NO3 excretion was prevented by either SP or adrenalectomy. The improvement in NO by MR antagonism was associated with an increase in renal endothelial nitric oxide synthase (eNOS) levels, as well as a shift from the inactive to active moieties of eNOS. That is, there was an increase in p-S1177 eNOS (active eNOS) and a decrease in p-T497 eNOS (inactive eNOS). Accordingly, restoration of NO may be one of the protective mechanisms of MR blockade.
In addition, the aforementioned studies found that the abnormalities in the ET system brought about by I/R-AKI are also MR-dependent. I/R-AKI increases the expression of ET. It also increases the ET receptor subtype-A (ETA) receptor (which leads to vasoconstriction) and had a variable effect on the ET receptor subtype-B (ETB) receptor (which causes NO-mediated vasodilation in the medulla). Thus, the enhanced levels of ET may activate the ETA receptor more than the offsetting ETB receptor during I/R-AKI, favoring vasoconstriction. Adrenalectomy and MR antagonism blunted the increases in ET and the ETA receptor, while increasing ETB receptor expression. Thus, abrogation of MR activation shifts the balance back in favor of the vasodilator actions of ETB and NO.
The Barrera-Chimal et al. study [18], not only demonstrated the efficacy of BR-4628 in I/R-AKI, but it also elucidated a novel mechanism by which MR activation leads to the imbalance in the ET/NO systems during I/R-AKI. Similar to the previous studies, they found that MR antagonism prevented I/R-AKI-induced reductions in p-S1177 eNOS, NO, and ETB receptor expression. Because aldosterone can inactivate ETB (and consequently eNOS activity) in cultured pulmonary cells by inducing oxidant-dependent sulfenic acid modification in its Cys-405 residue [20], they tested whether this mechanism accounts for the inactivation of the ETB receptor and the decreased NO observed during I/R-AKI. They found that I/R caused sulfenic acid modification of the ETB receptors. BR-4628 prevented this modification, which in turn restored NO generation and suppressed renal injury, thus explaining how MR activation during I/R-AKI leads to decreased activation of the ETB receptor, with a subsequent decrease in NO [18]. Moreover, in a subsequent study, they used tissue-specific MR knockout mice and demonstrated that it was the MR on the VSMC (not endothelial cells) that drives the injury response to cyclosporine [21]. Figure 1 summarizes the proposed mechanism by which MR antagonism may be beneficial against I/R-AKI. Acute I/R activates MRs on VSMC, which increases oxidative stress, thus leading to sulfenic acid modification of the ETB receptors, which render them inactivate. Inactivation of the ETB receptor reduces the active moiety of eNOS and consequently NO. Hence, ET, which is also increased during I/R-AKI, will predominantly act upon the ETA receptors and cause an unopposed ET-induced vasoconstriction, which may propagate renal hypoperfusion and thereby injury.

Mechanisms through which an ischemic insult results in renal injury: ischemia activates the MRs in the VSMC and ET-1. a An ischemic insult increases MR activation and ET-1 in the VSMC and ET cells respectively. ET-1 via ETA, increases ROS and vasoconstriction. Ischemia-mediated MR activation also increases ROS, which induces a cysteine-sulfenic acid modification (Cys-S-OH) of the ETB receptor; this causes a decrease in serine phosphorylated endothelial NO synthase (pS1777eNOS; active moiety) and thereby NO, further propagating the vasoconstriction and AKI. b Addition of an MR antagonist blocks the ischemia-mediated MR activation, thereby decreasing ROS generation. This prevents the Cys-S-OH modification of ETB, increasing the active moiety of eNOS and hence NO generation. This leads to vasodilation, which in turn may decrease renal injury.
Mechanisms through which an ischemic insult results in renal injury: ischemia activates the MRs in the VSMC and ET-1. a An ischemic insult increases MR activation and ET-1 in the VSMC and ET cells respectively. ET-1 via ETA, increases ROS and vasoconstriction. Ischemia-mediated MR activation also increases ROS, which induces a cysteine-sulfenic acid modification (Cys-S-OH) of the ETB receptor; this causes a decrease in serine phosphorylated endothelial NO synthase (pS1777eNOS; active moiety) and thereby NO, further propagating the vasoconstriction and AKI. b Addition of an MR antagonist blocks the ischemia-mediated MR activation, thereby decreasing ROS generation. This prevents the Cys-S-OH modification of ETB, increasing the active moiety of eNOS and hence NO generation. This leads to vasodilation, which in turn may decrease renal injury.
Conclusion
In conclusion, MR activation has been implicated as a key mechanism propagating ischemic AKI. It influences various facets of its pathophysiology, including its long-term sequelae. Hence, drugs blocking the MR may be potentially useful in AKI. Despite this promise, there has been limited interest in using these drugs clinically, in part because of the fear of side effects. The advent of newer nonsteroidal MR antagonists with similar benefits and limited adverse effects may stimulate interest in this class of drugs, particularly in those forms of AKI in which hypoperfusion plays a prominent role such as in postoperative patients and during the post-transplant period.
Disclosure Statement
The authors do not have any conflict of interests to declare.
References
Selected paper from a presentation at the 2016 AKI and CRRT UABUCSD O'Brien Center Symposium, San Diego, Calif., USA, February 16, 2016. This symposium was supported in part from a National Institutes of Health grant for the UAB-UCSD O'Brien Center for Acute Kidney Injury Research (P30 DK079337).
