

Abstract
Background: The rapid delayed rectifier K+ current (IKr), carried by the hERG protein, is one of the main repolarising currents in the human heart and a reduction of this current increases the risk of ventricular fibrillation. α1-adrenoceptors (α1-AR) activation reduces IKr but, despite the clear relationship between an increase in the sympathetic tone and arrhythmias, the mechanisms underlying the α1-AR regulation of the hERG channel are controversial. Thus, we aimed to investigate the mechanisms by which α1-AR stimulation regulates IKr. Methods: α1-adrenoceptors, hERG channels, auxiliary subunits minK and MIRP1, the non PIP2-interacting mutant D-hERG (with a deletion of the 883-894 amino acids) in the C-terminal and the non PKC-phosphorylable mutant N-terminal truncated-hERG (NTK-hERG) were transfected in HEK293 cells. Cell membranes were extracted by centrifugation and the different proteins were visualized by Western blot. Potassium currents were recorded by the patch-clamp technique. IKr was recorded in isolated feline cardiac myocytes. Results: Activation of the α1-AR reduces the amplitude of IhERG and IKr through a positive shift in the activation half voltage, which reduces the channel availability at physiological membrane potentials. The intracellular pathway connecting the α1-AR to the hERG channel in HEK293 cells includes activation of the Gαq protein, PLC activation and PIP2 hydrolysis, activation of PKC and direct phosphorylation of the hERG channel N-terminal. The PKC-mediated IKr channel phosphorylation and subsequent IKr reduction after α1-AR stimulation was corroborated in feline cardiac myocytes. Conclusions: These findings clarify the link between sympathetic nervous system hyperactivity and IKr reduction, one of the best characterized causes of torsades de pointes and ventricular fibrillation.
Introduction
The classical paradigm says that for an arrhythmia to be induced both a substrate and a trigger is needed. The human ether-a-go-go related gene (hERG) encodes the pore forming protein of the rapidly activated delayed rectifier K+ current (IKr), one of the most important currents that contributes to the repolarisation of the human heart. Reduction of this current either by a mutation in the gene or by a blockade of the channel by drugs produce congenital and acquired Long QT Syndrome (LQTS) respectively, creating a substrate that increases the risk of ventricular fibrillation [1]. The clinical observation showed that, in patients with LQTS, arrhythmias are typically triggered during physical or emotional stress, suggesting a link between sympathetic stimulation and arrhythmias. Sympathetic stimulation exerts different proarrhythmic effects, such as an increase of If or L-type Ca2+ currents and a reduction of hERG potassium channel activity [2,3]. In fact, activation of both β- and α-adrenoceptors (-ARs) regulate IKr.
β-AR stimulation has been proposed to regulate the Kv4.3 and hERG channels in cardiac myocytes [4] and heterologous systems inducing either an increase or a decrease of the hERG current. The proposed regulatory mechanisms of hERG included direct interaction of cAMP with the CNBD (cyclic nucleotide binding domain) of the channel, PKA-dependent phosphorylation and dynamic interaction with the 14-3-3 protein [5,6,7,8]. However, it was later demonstrated that cyclic nucleotides do not directly modulate hERG channels [9], and hERG-14-3-3 interaction has not been demonstrated in native cells. In fact, only an increase of IKr via the activation of PKA has been demonstrated in native ventricular cells [10]. Regarding α1 adrenoceptors, although α1-AR activation also reduces native IKr and IhERG in cardiac myocytes and in heterologous expression systems respectively, the specific mechanism remains elusive.
In 1999 it was reported for the first time a reduction in the IhERG upon α1-AR activation, but the G protein involved was not determined [11]. Later works proposed that phosphatidylinositol 4,5-bisphosphate (PIP2) consumption by PLC mediated hydrolysis might cause such reduction [12], probably involving a direct interaction between PIP2 and the C terminus of the channel [13]. However, the PIP2-dependent regulation of hERG channel has been questioned since and is currently believed it is not essential for normal channel function [14,15].
Simultaneously, other works proposed that the reduction of the IhERG upon α1-AR stimulation was due to the phosphorylation of the hERG channel by PKA and PKC [16,17,18]. However, when mutation experiments in Xenopus oocytes showed that deletion of most of the consensus phosphorylation sites did not prevent the effect, authors suggested that hERG channels were modulated by PKA and PKC independently of direct phosphorylation [19]. It is important to note that not all the putative phosphorylation sites were mutated in that work and, therefore, the direct channel phosphorylation cannot be totally excluded.
In this work we aimed to investigate the molecular mechanism by which sympathetic activation of α1-AR regulates ventricular IKr. Thus, we analyzed the effect of α1-AR stimulation on hERG channel in Human Embryonic Kidney (HEK293) cells and on IKr in feline cardiac myocytes.
Materials and Methods
Cell culture and transfection
Human Embryonic Kidney cells and a cell line stably expressing hERG channel [20] (HEK-hERG; Kindly provided by Dr. J. Hancox, University of Bristol, with the permission of Dr. C.T. January, University of Wisconsin) were cultured in DMEM (Dubelcco's modified Eagle's medium), supplemented with 10% of fetal bovine serum and 1% of penicillin-streptomycin-amphotericin cocktail. HEK-hERG grew in the presence of 50 µM of geneticin. Cell cultures were maintained in 5% CO2 at 37°C. 4 µg of D-hERG cDNA (kindly provided by Dr. T.V. McDonald, Albert Einstein College of Medicine, NY) or NTK-hERG cDNA (kindly provided by Dr. M.C. Sanguinetti, University of Utah) were transiently cotransfected with 1 µg of α1-adrenoceptor cDNA (kindly provided by Dr. C. Hague, University of Washington) along with 0.2 µg of GFP cDNA (kindly provided by Dr. M.T. Perez-Garcia, University of Valladolid) using Fugene 6 (Roche) following manufacturer instructions. Cells were used for electrophysiological studies and Western blot experiments 48-72 h after transfection.
Western blot
HEK293 cell membranes were isolated by centrifugation at 40000g, fractionated on 10 % SDS-polyacrylamide gels and transferred to nitrocellulose membranes (Amersham Biosciences). Nitrocellulose membranes were blocked in TTBS solution (Tris-HCl 50 mM pH 7.5, NaCl 150 mM, Tween-20 0.05%) containing BSA 3%. Blots were incubated with primary antibodies anti-hERG (1:200, Santa Cruz Biotechnology); anti-α1-AR (1:400; Oncogene Research Products); anti-minK (1:100; Santa Cruz Biotechnology); and anti-MIRP1 (1:200; Abnova). Secondary antibodies were conjugated with Horseradish Peroxidase and blots were developed using enhanced chemiluminescence (West Pico, Thermo Scientific).
Isolation of feline cardiomyocytes
Animal studies have been approved by the Ethics Committee for the Humane Use of Laboratory Animals of the University of Colima (México) and the experiments were conducted following the ethical standards laid down in the Declaration of Helsinki and its later amendments.
Single right ventricular myocytes were obtained from adult cats as described previously [21]. Hearts were mounted on a Langendorff apparatus and perfused for 5 min with normal Tyrode's solution, and then switched to a nominally calcium-free solution for an additional 5 min. Thereafter, the hearts were perfused for 30 min with a zero-calcium solution containing 1 mg/ml type I collagenase (Sigma-Aldrich, St. Louis, MO) and 0.05 mg/ml protease XIV (Sigma-Aldrich). The enzymes were washed out by perfusion with a high-potassium, low chloride saline (KB medium) for 5 min [22]. The free wall of the right ventricle was dissected away from the rest of the heart, placed in beakers, and cut into small pieces. Single cells were maintained in a high-potassium, low chloride solution at 4°C for 2 h before use in electrophysiological experiments.
Tyrode's solution had the following composition (in mM): 125 NaCl, 24 NaHCO3, 0.42 NaH2PO4, 5.4 KCl, 1.8 CaCl2, 1.05 MgCl2, 11 glucose, and 10 taurine. The solution was equilibrated with 95% O2 and 5% CO2, pH 7.4. Nominally calcium-free solution was prepared by omitting CaCl2 from the Tyrode's solution. The high-potassium, low-chloride KB medium had the following composition (in mM): 80 potassium glutamate, 40 KCl, 20 taurine, 3 KH2PO4, 10 glucose, 10 HEPES, and 0.2 EGTA. pH 7.4 with KOH.
Patch-clamp recordings
Macroscopic currents were recorded at room temperature (22°C) in the whole-cell configurations of the patch-clamp technique [23], using Axopatch-200B amplifiers (Molecular Devices). Borosilicate capillary glass were pulled obtaining a tip resistance of 1-3 MΩ after filled with the internal solution. For HEK293 cells the internal solution was (in mM) 125 KCl, 5 MgCl2, 5 EGTA-K, 10 HEPES-K and 5 Na-ATP adjusted to pH 7.2 with KOH. For cardiac myocytes the internal solution was (in mM): 90 potassium aspartate, 54 KCl, 10 KH2PO4, 5 HEPES, 0.1 EGTA, pH 7.3 with KOH.
Following the patch rupture, whole cell membrane capacitances were measured from integration of the capacitive transients elicited by voltage steps from -50 to -60 mV, which did not activate any time dependent membrane current. Series resistances were compensated 80% in order to minimize voltage errors and were checked regularly throughout the experiment to ensure that there were no variations with time. The voltage-clamp experimental protocols were controlled with the “Clampex” program of the “pClamp” software (Molecular Devices). HEK293 cells were perfused with the external solution containing (in mM): 136 NaCl, 4 KCl, 1 MgCl2, 10 HEPES-Na, 1.8 CaCl2 and 10 glucose, pH 7.4 with NaOH. For cardiac myocytes the bathing solution was (in mM): 140 NaCl, 5.4 KCl, 0.1 CaCl2, 0.4 CoCl2, 1.0 MgCl2, 10 HEPES and 11 glucose, pH adjusted to 7.4 with NaOH. HMR-1556 1 µM was added to block IKs. For HEK-hERG cells terfenadine 1 µM was added at the end of the experiment and the remaining endogenous current was subtracted.
To elicit IhERG and IKr we applied depolarizing pulses from -40 to +70 in 10 mV increments for 2 s every 30 seconds from a holding potential of -80 mV. These pulses were followed by a pulse at -40 mV for 5 s to measure tail current. I-V curves were obtained by measuring the current at the end of the test pulses, normalized to cell capacitance and expressed as pA/pF. The peak of the tail current was normalized to the maximum current and curves were fitted to Boltzmann relation I=1/[1 + e(Vh-V)/k], were I is the amplitude of the tail current, Vh is a half-maximal activation, V is the applied membrane voltage and k is the slope factor. Clampfit 10.2 software was used for the analysis of currents. The deactivation kinetics were calculated by fitting the decay of tail currents to a monoexponential function.
Drugs
Phenylephrine (PE), propranolol and neomycin were from Sigma-Aldrich. GP antagonist 2A (GPant2A); and bisindolylmaleimide 1 (BIS-1) were from Calbiochem. 1-phosphatidyl-1D-myo-inositol-3,4-bisphosphate (diC8-PIP2) was from Echelon Biosciences. Anti-PIP2 was from Santa Cruz Biotechnology. HMR-1556 was from Tocris.
Statistical analysis
Values are presented as mean ± SEM. Student's t-test for paired data was used to compare the difference between two means. p<0.05 was considered significant.
Results
Phenylephrine reduces IhERG in HEK293 cells
Cell lysates were immunoblotted to confirm the membrane expression of the α1A-adrenergic receptor and the hERG channel. The adrenoceptor, as well as the two bands, mature glycosilated and inmature non-glycosilated, characteristic of the hERG channel proteins were detected only in transfected HEK-hERG cells (Fig. 1A).
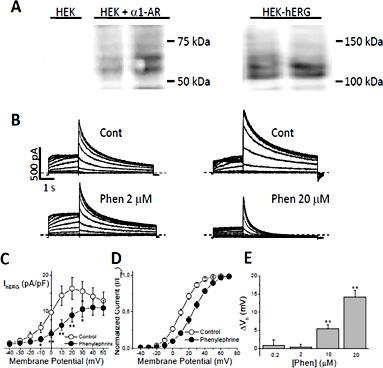
α1-AR stimulation reduces IhERG and shifts the activation half voltage. A) Membrane expression of α1A-ARs in untransfected and transfected HEK293 cells (left) and channel expression in HEK293 cells stably expressing hERG (right); duplicates are shown. B) Representative hERG current recordings in two HEK-hERG cells transfected with the α1-adrenoceptor before (Cont) and after administration of 2 or 20 µM phenylephrine (Phen). C) Current density-voltage curve and D) voltage dependence of activation in control and after α1A-AR stimulation. E) Concentration-dependence of the effect of phenylephrine on the voltage dependence of IhERG activation. The displacement of the activation half voltage (ΔVh) is plotted against phenylephrine concentration. Mean ± SEM; n = 7-37 paired cells. *p < 0.05; **p < 0.01. Dashed lines indicate the zero current level.
α1-AR stimulation reduces IhERG and shifts the activation half voltage. A) Membrane expression of α1A-ARs in untransfected and transfected HEK293 cells (left) and channel expression in HEK293 cells stably expressing hERG (right); duplicates are shown. B) Representative hERG current recordings in two HEK-hERG cells transfected with the α1-adrenoceptor before (Cont) and after administration of 2 or 20 µM phenylephrine (Phen). C) Current density-voltage curve and D) voltage dependence of activation in control and after α1A-AR stimulation. E) Concentration-dependence of the effect of phenylephrine on the voltage dependence of IhERG activation. The displacement of the activation half voltage (ΔVh) is plotted against phenylephrine concentration. Mean ± SEM; n = 7-37 paired cells. *p < 0.05; **p < 0.01. Dashed lines indicate the zero current level.
Transfection was functional and cells responded to receptor stimulation with phenylephrine (PE). Thus, hERG channel current was recorded before and five minutes after 20 µM PE administration and a reduction on both activated IhERG and tail current was observed (Fig. 1B). The current-voltage (I-V) relationship normalized to cell capacitance of the IhERG at the end of the 2 seconds pulse in control and PE stimulation shows current inhibition in a wide voltage range: between -10 to +30 mV. Because of the channel rectification properties, as the voltage increased the contribution of hERG channels to the total current become smaller and therefore the effect of PE was lower (Fig. 1C). Analysis of hERG tail currents showed that PE accelerates the current deactivation. When fitted to a monoexponential function, τdeact was accelerated from 1331 ± 74 to 639 ± 44 ms (n = 37 paired cells, p<0.0001). The voltage dependence of hERG activation was calculated from plots of tail current amplitudes (normalized to the peak current) versus voltage. PE administration caused a 14 mV positive shift in the activation half voltage (Vh), from 10.9 ± 1.9 to 25.1 ± 2.1 mV (n = 37 paired cells; p < 0.001), and this effect was concentration-dependent, with an EC50 = 12 µM (Fig. 1D, E). PE concentrations higher than 40 µM PE fully eliminated the IhERG.
α1-AR stimulation shifts hERG channel activation Vh
Since hERG channel has a great susceptibility to be blocked by many different drugs, we first tested whether the effects of PE on IhERG were due to α1-AR stimulation or to a blockade of the channel. Thus, when 20 µM PE was added to hERG expressing HEK cells not transfected with the receptor, we still observed a faster tail current deactivation (1420±98 vs 998±85 ms; n = 18 paired cells, p<0.0001), suggesting a direct open channel blockade of the channel by PE. Conversely, the activation half voltage was not affected by phenylephrine in the absence of the receptor (Fig. 2). This result demonstrates that the positive shift in the hERG activation half voltage requires α1-AR stimulation. In conclusion, the mechanism responsible for the IhERG reduction after the α1-AR activation is the positive displacement of the activation half voltage because, at a given membrane potential, the current amplitude is expected to be reduced.

minK and MIRP1 are not required for α1-AR induced regulation of IhERG. A) hERG Current recordings in a HEK-hERG cell not transfected with the α1A-adrenoceptor before (Cont) and after adding 20 µM phenylephrine (Phen). B) Western blot of minK and MIRP1 in untransfected (Ø) and transfected HEK-hERG cells. C) Displacement of the IhERG activation half voltage (ΔVh) induced by 20 µM phenylephrine in HEK-hERG cells not transfected with α1-AR (NO α1-AR) and cotransfected with α1-AR plus either minK or MIRP1. Mean ± SEM. n = 18, 7, 13 paired cells. *p<0.01. Dashed lines indicate the zero current level.
minK and MIRP1 are not required for α1-AR induced regulation of IhERG. A) hERG Current recordings in a HEK-hERG cell not transfected with the α1A-adrenoceptor before (Cont) and after adding 20 µM phenylephrine (Phen). B) Western blot of minK and MIRP1 in untransfected (Ø) and transfected HEK-hERG cells. C) Displacement of the IhERG activation half voltage (ΔVh) induced by 20 µM phenylephrine in HEK-hERG cells not transfected with α1-AR (NO α1-AR) and cotransfected with α1-AR plus either minK or MIRP1. Mean ± SEM. n = 18, 7, 13 paired cells. *p<0.01. Dashed lines indicate the zero current level.
Neither minK nor MIRP1 modulate the α1-AR regulation of hERG channels
Although the real hERG partners in native systems is a matter of debate, in heterologous expression systems hERG channels can interact with the accessory proteins minK and MIRP1 [24]. We wondered whether these proteins could affect the α1-AR-induced IhERG reduction, so we transfected HEK-hERG cells with the receptor plus one of the accessory subunits and tested the effects of PE. Membrane expression of minK and MIRP1 was confirmed by Western blot (Fig. 2B).
Current recordings showed that 20 µM PE shifted the IhERG+minK activation Vh to a similar extent than in the absence of the accessory protein (from 14 ± 4.1 to 27.7 ± 3.1 mV; n = 7 paired cells; p < 0.01). As expected, MIRP1 shifted hERG activation Vh to more negative potentials. However, when PE was added to the cells the approximately 14 mV shift in IhERG+MIRP1 paired cells; p < 0.01). As the effect of PE on the activation Vh of IhERG+minK, IhERG+MIRP1 and IhERG is basically identical, we can conclude that minK and MIRP1 do not alter the hERG channel response to the α1-ARs stimulation (Fig. 2C).
Intracellular pathway mediating the α1-AR modulation of hERG
The α1-adrenoceptor belongs to a G-protein coupled receptors family classically associated with Gαq dissociation, phospholipase C (PLC) activation, PIP2 hydrolysis and PKC activation. The involvement of the Gαq protein in the PE-induced IhERG reduction was confirmed with 50 µM of the specific Gαq blocker GPant2A (RPKPQQDWFDWDWM). In the presence of this blocker PE did not shift the activation half voltage of IhERG (Fig. 3), suggesting that αq is the relevant G protein in this pathway.

α1-AR induced IhERG regulation depends on PLC and PKC activation. A) hERG current recordings in a HEK-hERG cell transfected with the α1A-adrenoceptor in control (Cont) and after administration of 20 µM phenylephrine (Phen) in the presence of an anti-PIP2 antibody in the internal solution. B) Displacement of the IhERG activation half voltage (ΔVh) caused by α1-AR stimulation in cells with: the Gq protein blocker GPantagonist 2A 50 µM; the PLC inhibitors Neomycin 1 mM, anti-PIP2 antibody 80 nM and diC8-PIP2 10 µM; and the PKC blocker bisindolylmaleimide 1 (BIS-1) 10 µM. Mean ± SEM. n = 9, 9, 8, 11, 18 paired cells. Dashed lines indicate the zero current level.
α1-AR induced IhERG regulation depends on PLC and PKC activation. A) hERG current recordings in a HEK-hERG cell transfected with the α1A-adrenoceptor in control (Cont) and after administration of 20 µM phenylephrine (Phen) in the presence of an anti-PIP2 antibody in the internal solution. B) Displacement of the IhERG activation half voltage (ΔVh) caused by α1-AR stimulation in cells with: the Gq protein blocker GPantagonist 2A 50 µM; the PLC inhibitors Neomycin 1 mM, anti-PIP2 antibody 80 nM and diC8-PIP2 10 µM; and the PKC blocker bisindolylmaleimide 1 (BIS-1) 10 µM. Mean ± SEM. n = 9, 9, 8, 11, 18 paired cells. Dashed lines indicate the zero current level.
In the canonical pathway, Gαq protein-activated PLC hydrolyses PIP2 producing the PKC activators 1,2-diacylglycerol (DAG) and inositol-1,4,5,-trisphosphate (IP3). We blocked these steps with three chemical tools: neomycin and an anti-PIP2 antibody, which block the access of PLC to PIP2; and the non-hydrolysable PIP2 analogue diC8-PIP2, a putative competitive inhibitor of phospholipases [25,26,27,28]. The presence of each of the inhibitors prevented the PE-induced shift in IhERG activation half voltage (Fig. 3), indicating the involvement of PLC.
hERG channels are modulated by Ser/Thr kinases like PKA and PKC and by Tyrosine kinases such as Src [19,29,30]. Scansite database reveals 1 putative phosphorylation site for Src (Y1009) and 18 sites for PKA and/or PKC in the channel sequence. Our results showed that the effect of α1-AR stimulation on IhERG does not depend on tyrosine kinases mediated phosphorylation. In the presence of the Tyr-kinase inhibitor genistein 50 µM the activation Vh moved from 10.3 ± 3.4 to 25.9 ± 3 mV after adding phenylephrine (n = 9; p<0.001). However, when PKC was inhibited by BIS-1 (10 µM) PE was not able to shift the activation half voltage of IhERG to more positive potentials (from 11.2 ± 3.1 to 11.3 ± 3 mV; n=18 paired cells, ns; Fig. 3). Thus, PKC has a key role in the regulation of the hERG channel after α1-AR stimulation.
To confirm that PIP2 does not directly affect the channel modulation, HEK293 cells were cotransfected with the α1-AR plus the D-hERG mutant. This mutant has a deletion of the 883-894 amino acids in the C-terminal, the main site of interaction between PIP2 and hERG channels [13]. Perfusion of the cells with 20 µM PE shifted the activation half voltage of ID-hERG to more depolarized potentials (from 8.2 ± 3 to 20.1 ± 3 mV; n=11 paired cells; p <0.001; Fig. 4), same as was observed with the wild type channel. On the contrary, addition of the non-hydrolysable analogue diC-8-PIP2 (10 µM) or the PKC blocker BIS-1 (10 µM) prevented the effect of PE on the deletion mutant, supporting the role of PKC-dependent phosphorylation of the channel (Fig. 4C).

PIP2 interaction with hERG channel is not relevant for regulation by α1-AR A) Expression of the presumably non PIP2 interacting mutant D-hERG in transfected HEK293 cells (duplicates are shown). B) ID-hERG recordings in a HEK cell coexpressing the D-hERG and the α1-AR, before (Cont) and after 20 µM phenylephrine (Phen) administration. C) Displacement of the ID-hERG activation half voltage (ΔVh) caused by PE in cells without (Ø) and with diC8-PIP2 or bisindolylmaleimide 1. Mean ± SEM. n = 11, 7, 8 paired cells. *p<0.01. Dashed lines indicate the zero current level.
PIP2 interaction with hERG channel is not relevant for regulation by α1-AR A) Expression of the presumably non PIP2 interacting mutant D-hERG in transfected HEK293 cells (duplicates are shown). B) ID-hERG recordings in a HEK cell coexpressing the D-hERG and the α1-AR, before (Cont) and after 20 µM phenylephrine (Phen) administration. C) Displacement of the ID-hERG activation half voltage (ΔVh) caused by PE in cells without (Ø) and with diC8-PIP2 or bisindolylmaleimide 1. Mean ± SEM. n = 11, 7, 8 paired cells. *p<0.01. Dashed lines indicate the zero current level.
In previous works the four PKA phosphorylation sites and 17 of the 18 consensus PKC phosphorylation sites in hERG were mutated to alanine and the channel modulation by the α1-AR was similar to WT channels [19,31]. However, the T74, located in the Per-Arnt-Sim (PAS) domain of the channel, cannot be deleted or mutated to alanine without the loss of function of the channel, showing that this is a key residue in the channel function. Interestingly, the T74 was demonstrated to be phosphorylated by PKC [32]. So we then cotransfected HEK293 cells with the α1-AR and the NTK-hERG channel, a mutant whose 2-354 amino acids containing the PAS domain and the T74 have been removed [33]. Although both activation and deactivation kinetics are faster in NTK-hERG than in WT hERG, the activation half voltage is similar and was not regulated by PE (from 9.5 ± 4.4 to 11.3 ± 3.4 mV; n=10, ns). The absence of effect of α1-AR stimulation on the NTK-hERG activation Vh is again consistent with a major role of PKC in this regulation pathway. As expected, diC8-PIP2 or BIS-1 did not affect the INTKhERG activation Vh after PE exposure (Fig. 5).

hERG regulation by α1-ARs is dependent on PKC phosphorylation. A) Expression of the non PKC-phosphorylable mutant NTK-hERG in transfected HEK293 cells. B) INTK-hERG recordings in a HEK cell coexpressing the NTK-hERG and the α1-AR, before (Cont) and after 20 µM phenylephrine (Phen) administration. Since NTK-hERG tail deactivation kinetics are faster than WT, the inset shows an amplification of the tail currents. C) Displacement of the INTK-hERG activation half voltage (ΔVh) caused by 20 µM phenylephrine in cells without (Ø) and with diC8-PIP2 or bisindolylmaleimide 1 (BIS-1). Mean ± SEM. n = 10, 6, 7 paired cells. Dashed lines indicate the zero current level.
hERG regulation by α1-ARs is dependent on PKC phosphorylation. A) Expression of the non PKC-phosphorylable mutant NTK-hERG in transfected HEK293 cells. B) INTK-hERG recordings in a HEK cell coexpressing the NTK-hERG and the α1-AR, before (Cont) and after 20 µM phenylephrine (Phen) administration. Since NTK-hERG tail deactivation kinetics are faster than WT, the inset shows an amplification of the tail currents. C) Displacement of the INTK-hERG activation half voltage (ΔVh) caused by 20 µM phenylephrine in cells without (Ø) and with diC8-PIP2 or bisindolylmaleimide 1 (BIS-1). Mean ± SEM. n = 10, 6, 7 paired cells. Dashed lines indicate the zero current level.
α1-Adrenergic modulation of IKr in feline cardiac myocytes
In order to test whether the results obtained in HEK-hERG cells could be extrapolated to native cardiac cells we used isolated feline ventricular cardiomyocytes. All the experiments with ventricular myocytes were performed in the presence of the β-blocker propranolol 100 nM to avoid any potential effect of PE on β-ARs. As previously observed in transfected HEK293 cells, in feline native cardiomyocytes the stimulation of the α1-AR with PE reduced the IKr activated current and shifted the activation half voltage to more depolarized potentials (from -7.4 ± 1.1 to -1.1 ± 1.2 mV, n=10 paired cells; p < 0.01). The specific α1-AR blocker prazosin (1 µM) abolished the effect on IKr indicating that is dependent on receptor activation (Fig. 6). The relevance of PKC in the intracellular pathway was tested using diC-8-PIP2 and BIS-1 and, as expected, they abolished the PE effect on IKr. Therefore, PKC has a critical role on IKr regulation upon α1-AR stimulation in ventricular myocytes.
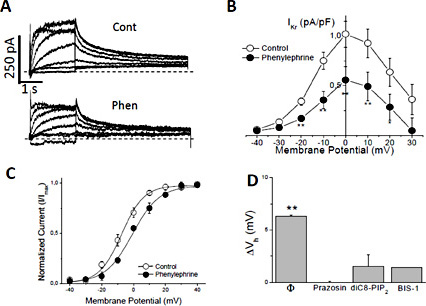
α1-AR stimulation reduces IKr by a PKC-phosphorylation mediated mechanism. A) IKr recordings in a feline ventricular myocyte in control (Cont) and after application of 20 µM phenylephrine (Phen). B) Current density-voltage curve and C) voltage dependence of activation before and after α1-AR stimulation. D) Displacement of the IKr activation half voltage (ΔVh) induced by α1-AR stimulation in cells without (Ø) and with prazosin (1 µM), diC8-PIP2 or bisindolylmaleimide 1 (BIS-1). Mean ± SEM. n = 10, 5, 6, 5 paired cells. * p < 0.05; **p < 0.01. Dashed lines indicate the zero current level.
α1-AR stimulation reduces IKr by a PKC-phosphorylation mediated mechanism. A) IKr recordings in a feline ventricular myocyte in control (Cont) and after application of 20 µM phenylephrine (Phen). B) Current density-voltage curve and C) voltage dependence of activation before and after α1-AR stimulation. D) Displacement of the IKr activation half voltage (ΔVh) induced by α1-AR stimulation in cells without (Ø) and with prazosin (1 µM), diC8-PIP2 or bisindolylmaleimide 1 (BIS-1). Mean ± SEM. n = 10, 5, 6, 5 paired cells. * p < 0.05; **p < 0.01. Dashed lines indicate the zero current level.
Discussion
Mechanisms of IhERG reduction upon α1-AR stimulation
In this work, the activation of the α1-AR with phenylephrine reduces the IhERG amplitude as previously published [12,13,19,31,34]. The experiments performed in hERG-HEK cells not expressing α1-AR show that the shift in the activation Vh to more positive potentials is the cause of IhERG reduction upon α1A-AR stimulation, because there will be fewer available channels at any physiological membrane potential. The regulatory proteins minK and MIRP1 are not relevant for the α1A-adrenergic regulation of IhERG.
Jiang et al. found for the first time a reduction on IhERG amplitude by α1A-AR stimulation; however the authors did not identify the G protein involved [11]. Although this receptor can activate Gαs protein and the cAMP/PKA pathway in cardiac myocytes [35], the most extensively documented biochemical responses to α1-AR stimulation in the heart is Gαq protein activation of phospholipase C-mediated hydrolysis of PIP2 that produces DAG and IP3 and leads to PKC activation by these second messengers [36,37,38]. Our results show that the regulation of hERG current is mediated by the canonical Gαq-PLC intracellular signalling pathway. It has been proposed that the effect of PLC activation on IhERG upon α1-AR stimulation might be caused by PIP2 depletion and loss of PIP2-hERG interaction [12,13]. However, other authors suggest that the channel modulation lies on DAG production and PKC-dependent phosphorylation [19,31]. As will be discussed below, our experiments with deletion mutants support the latter.
Implication of Protein Kinases
The most common mechanism to regulate K+ channels is reversible phosphorylation [35,39]. The Tyrosine kinase Src shifts the activation half voltage of both IKr and IhERG [30,40]. In our work, however, inhibition of Tyrosine kinases did not prevent the shift in the activation Vh induced by α1-AR stimulation, suggesting that they are not involved in this regulatory pathway.
Although several works propose a role for PKC in the modulation of hERG channels in response to Gαq protein coupled receptors [19,29,32,41], one paper using the PKC inhibitor chelerythrine excluded PKC involvement in α1-AR modulation of hERG [12]. In that study cells were incubated extracellularly for one hour with a chelerythrine concentration barely above the IC50, so probably neither the incubation time nor the concentration used were enough to cause a significant blockade. We have administrated the PKC blocker bisindolylmaleimide-1 in the intracellular solution to ensure inhibition of the kinase and then found that α1-AR stimulation does not shift the activation half voltage of IhERG. Thus, PKC has a key role in the regulation of the hERG channel after α1-AR stimulation.
Thomas et al. suggested that α1-AR regulation of hERG channel was mediated by both PKA and PKC by a mechanism independent of direct channel phosphorylation [19], since PKA inhibition prevented the effect of α1-AR stimulation on IhERG but mutation of the four PKA phosphorylation sites did not. It is currently accepted that KT5720, the PKA blocker used, can also inhibit PKC at physiological ATP concentrations [42] and therefore involvement of PKA is not sustained. That work also ruled out the direct phosphorylation of the channel by PKC because mutation of 17 out of 18 phosphorylation sites to alanine did not prevent the α1-AR effect on hERG [19]. Authors could not exclude the T74 site, located in the PAS domain, as the target for PKC-dependent phosphorylation of hERG channel after α1-AR stimulation because mutation of that residue resulted in non-functional channels. In this sense, it is currently known that PKC modulates hERG channels [29,31,41,43] precisely by direct phosphorylation of the T74 site [32]. Whereas point mutation or deletion of T74 causes channel loss of activity [19,32], the NTK-hERG mutant lacks the full PAS domain and is functional enough to allow exploring the modulation by PKC [32,33]. In the present work, α1-ARs stimulation does not modulate INTK-hERG, supporting a role for direct channel phosphorylation by PKC.
PIP2 regulates inward rectifier potassium channels [44,45,46,47,48,49,50], voltage dependent Ca2+ channels [reviewed in 51], cardiac Na+/Ca2+ exchanger [52], KCNQ1 [53] and a role for PIP2 in the regulation of IKr/IhERG by α1-AR has been proposed [12,13,54].
Negative phosphate groups of the PIP2 head interact with positively charged amino acids of the channel proteins [45,55] and the deletion or substitution of the 883-894 amino acids in the hERG channel C-terminal eliminates the apparent main interaction site between PIP2 and hERG [13]. We used the D-hERG mutant (deletion of C-terminal 883-894 amino acids) in order to test whether PIP2 might be involved in the α1-AR regulation of IhERG. D-hERG mutant responds to α1-AR stimulation and behaves like the WT despite the lack of the apparent PIP2 interaction site. On the contrary, PKC inhibition fully abolishes α1-adrenergic modulation of D-hERG, which is again consistent with a relevant role of direct phosphorylation by PKC in this pathway.
Limitations of the study
Although PE induces a positive shift in the activation Vh in both HEK cells and feline cardiac myocytes, the smaller effect of PE in feline myocytes can be explained by different factors such as a lower α1-AR expression. Feline myocytes are native cells, whereas in HEK cells the receptor was transfected and overexpressed. It might also be differences in efficiency of coupling between the components of the intracellular pathway (receptor-Gq proteins; Gq-PLC; PLC-PKC;…) or a different stoichiometry between PKC and ERG channels.
Feline IKr is not identical to transfected IhERG. In addition to the possible differences between feline and human ERG channel, it is well accepted that hERG channel resembles most but not all the characteristics of human IKr and that accessory subunits would be required to create the chanelosome responsible for native IKr. However, we found that the main candidates, minK or MIRP1, do not modify the effect of Phenylephrine on hERG.
Translational perspective
Our results in native, feline, cardiomyocytes reproduce what was observed in transfected cells. Thus, α1-AR stimulation reduces the IKr activated and tail currents by shifting the voltage-dependence of activation to more depolarized potentials in a mechanism that requires PLC and PKC activation and PKC-dependent channel phosphorylation.
One common trigger of arrhythmia in LQT2 patients seems to be emotional stress and high and unexpected noise [56]. Symptomatic patients that suffer lethal and no lethal cardiac events are related with an increase in the sympathetic tone [3,57,58,59], which means a clear relation between sympathetic nervous system and arrhythmias. In fact, auditory stimulus-induced arrhythmias are almost exclusively associated with mutations in KCNH2, the gene encoding hERG [2]. β-AR and α-AR are activated by noradrenalin, but although β-ARs are saturated by moderate sympathetic activation, α1-ARs activate in situations of sympathetic hyperactivity such as emotional or physical stress. In this sense, an anti-arrhythmic effect of α1-AR blockade has been demonstrated in an animal model of LQT2 [60]. Here, we clarified the link between sympathetic nervous system hyperactivity and IKr reduction, one of the best characterized causes of torsades de pointes and ventricular fibrillation.
Acknowledgements
This work was supported by Universidad del País Vasco UPV/EHU grants (PPM12/12 and PES12-33); Basque Government grant IT653-13 and MINECO (SAF2013-46708-R) to O.C.; and UPV/EHU grant (EHUA12/12) to M.G. The authors wish to thank Dr. E. Medei, from the University of Rio de Janeiro in Brasil, and Dr. A.A. Rodríguez-Menchaca, from the Universidad Autónoma de San Luis Potosí in México for the useful comments and critical review of the manuscript.
Disclosure Statement
The authors have nothing to disclose.

