

Abstract
Background: Nuclear factor erythroid 2-related factor 2 (NRF2) is a critical transcription factor for the antioxidative stress response and it activates a variety of cytoprotective genes related to redox and detoxification. NRF2 activity is regulated by the oxidative-stress sensor molecule Kelch-like ECH-associated protein 1 (KEAP1) that induces proteasomal degradation of NRF2 through ubiquitinating NRF2 under unstressed conditions. Because oxidative stress is a major pathogenic and aggravating factor for kidney diseases, the KEAP1-NRF2 system has been proposed to be a therapeutic target for renal protection. Summary: Oxidative-stress molecules, such as reactive oxygen species, accumulate in the kidneys of animal models for acute kidney injury (AKI), in which NRF2 is transiently and slightly activated. Genetic or pharmacological enhancement of NRF2 activity in the renal tubules significantly ameliorates damage related to AKI and prevents AKI progression to chronic kidney disease (CKD) by reducing oxidative stress. These beneficial effects of NRF2 activation highlight the KEAP1-NRF2 system as an important target for kidney disease treatment. However, a phase-3 clinical trial of a KEAP1 inhibitor for patients with stage 4 CKD and type-2 diabetes mellitus (T2DM) was terminated due to the occurrence of cardiovascular events. Because recent basic studies have accumulated positive effects of KEAP1 inhibitors in moderate stages of CKD, phase-2 trials have been restarted. The data from the ongoing projects demonstrate that a KEAP1 inhibitor improves the glomerular filtration rate in patients with stage 3 CKD and T2DM without safety concerns. Key Message: The KEAP1-NRF2 system is one of the most promising therapeutic targets for kidney disease, and KEAP1 inhibitors could be part of critical therapies for kidney disease.
Introduction
The respiratory system continuously acquires oxygen for the production of cellular energy via intracellular aerobic respiration in mitochondria. During energy production, reactive oxygen species (ROS) are generated simultaneously. These molecules are harmful to living cells due to their high reactive nature [1]. Electrophiles, which contain centers with low electron densities, arise in cells as a consequence of both physiological and pathological processes and may, in turn, react with critical biomolecules, including DNA. Excessive electrophiles damage cells in the form of oxidative stress. Recent studies have demonstrated that oxidative stress is involved in the pathogenesis and progression of diseases and is considered to be a final common pathway in a variety of diseases [2].
To maintain cellular redox homeostasis and to avoid harmful oxidative conditions, the endogenous defense system, which consists of Kelch-like ECH-associated protein 1 (KEAP1) and nuclear factor erythroid 2-related factor 2 (NRF2), regulates the expression of genes related to detoxification in an electrophile-dependent manner [3]. Previous investigations have demonstrated the protective effects of NRF2 against the development and aggravation of various diseases [4]. Furthermore, clinical trials of NRF2 activators have been conducted. For example, studies have shown that the activation of NRF2 exerts cytoprotective effects by enhancing anti-inflammatory and/or antioxidative stress responses in animal models of respiratory disease [5,6], diabetes mellitus [7,8], sickle-cell disease [9] and endotoxin shock [10]. Additionally, phase-3 clinical trials (e.g., the DEFINE and CONFIRM studies) reported that the NRF2 activator dimethyl fumarate (DMF) suppresses the progression and recurrence of multiple sclerosis, which is a progressive neurodegenerative disease caused by ROS-mediated demyelination [11,12]. Based on these positive results, DMF is now commercially available as Tecfidera® for therapeutic use against multiple sclerosis. Moreover, clinical trials of another NRF2 activator, 1-(2-cyano-3-,12-dioxooleana-1,9(11)-dien-28-oyl)-methyl ester (CDDO-methyl ester, also known as RTA 402) are currently being conducted for the treatment of pulmonary hypertension (https://clinicaltrials.gov/ct2/show/NCT02036970) and connective tissue disease-associated pulmonary arterial hypertension (https://clinicaltrials.gov/ct2/show/NCT02657356).
Kidney diseases are also thought to be related to oxidative stress [2,13,14], and basic research has generated evidence that NRF2 activation prevents kidney disease progression by protecting cells from oxidative damage. Therefore, there is a need for NRF2-activating medicines to protect against kidney diseases, for which there are currently no effective medications. Here, we summarize basic studies of NRF2 function in kidney diseases and clinical trials of NRF2 activators for kidney disease treatment.
Oxidative Stress and Kidney Diseases
Acute kidney injury (AKI) is a syndrome that involves a rapid reduction of renal function within days or weeks and causes high morbidity and mortality [15]. Recently, AKI has been considered to be one of the major risk factors causing chronic kidney disease (CKD) and end-stage renal disease (ESRD), which require expensive renal-replacement therapies, such as hemodialysis and peritoneal dialysis [16,17]. Additionally, CKD and ESRD are closely linked to life-threatening diseases, including cerebrovascular and cardiovascular diseases [18,19]. Therefore, preventing the progression of AKI is an emergent and important medical issue for achieving better outcomes and reducing costs.
Basic research has shown that oxidative stress is a major aggravating factor for the pathogenesis and progression of kidney diseases [20,21,22]. Low-level ROS function as signaling molecules for cellular proliferation and vascular homeostasis under healthy conditions [14]; however, ROS accumulate massively in the kidneys due to pathological conditions caused by angiotensin II signaling [23,24], a high-salt diet [25], hypertension [23,26], inflammation [27], hyperglycemia [28] or nephrotoxic drugs [29].
Clinically, the induction of ROS generation by ischemia reperfusion injury (IRI) is a major cause of AKI through oxidative stress-mediated damage in the kidney [20]. During the ischemic phase of IRI, low levels of ROS are produced in the mitochondria [30,31]. During the subsequent reperfusion phases of IRI, ROS are generated rapidly by mitochondria, nicotinamide adenine dinucleotide phosphate (NADPH) oxidases, or inflammatory cells and they give rise to strong oxidative stress [14,31]. Therefore, eliminating oxidative stress has been considered to be an important therapeutic strategy for treating both the pathogenesis and progression of kidney diseases.
The KEAP1-NRF2 System
To protect cells from harmful oxidative stress, the KEAP1-NRF2 system allows the cell to sense and respond to oxidative stress conditions [3]. NRF2 is a master transcriptional regulator for genes related to redox status and antioxidant effects. In healthy cells, NRF2 is captured by KEAP1, which is a subunit protein of the E3-ubiquitin ligase specific for NRF2, and is degraded through the ubiquitin-proteasome pathway (Fig. 1) [32]. Electrophiles emerge under oxidative stress conditions and adduct to specific cysteine residue (Cys) of KEAP1. This cysteine modification alters the structure of KEAP1 to inhibit KEAP1-NRF2 binding. More than 10 sensor Cys have been identified in KEAP1, each of which reacts to a variety of electrophiles in different ways to sense many types of oxidative stress [33]. NRF2 molecules that escape from KEAP1-mediated capture and ubiquitination accumulate in the nucleus of cells exposed to oxidative stress and form a heterodimer with small MAF (sMAF) proteins to bind a specific recognition sequence, the CNC-sMAF binding element (CsMBE; 5′-[A/G]TGA[G/C]nnnGC-3′), which is also known as an antioxidant- or electrophile-responsive element [34,35]. Functional CsMBEs are localized in regulatory regions of genes that encode antioxidants, detoxification enzymes, glutathione synthetic enzymes, metabolic enzymes, and heme metabolic enzymes and in genes involved in mitochondrial function (Fig. 1) [36,37,38].
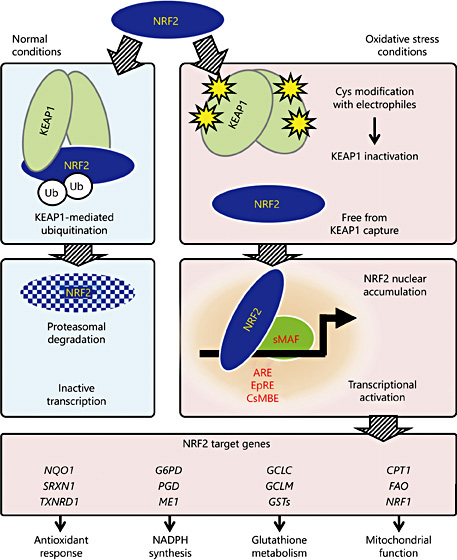
Molecular mechanism of the KEAP1-NRF2 system during the oxidative stress response. NRF2 is degraded and inactivated via ubiquitin-proteasome-dependent degradation after being captured by the KEAP1 homodimer in healthy cells (left). Electrophiles emerge during oxidative stress and alter the conformation of KEAP1 by directly adducting to the specific sensor cysteine residues (Cys) in KEAP1. Because this modification inactivates the interaction between KEAP1 and NRF2, NRF2 avoids degradation in cells exposed to oxidative stress. The stabilized NRF2 translocates into the nucleus, where NRF2 activates the transcription of its target genes by binding to NRF2 recognition sequences (i.e., ARE, EpRE, or CsMBE) as NRF2-sMAF heterodimers. Representative NRF2 target genes are separately listed in 4 categories (below).
Molecular mechanism of the KEAP1-NRF2 system during the oxidative stress response. NRF2 is degraded and inactivated via ubiquitin-proteasome-dependent degradation after being captured by the KEAP1 homodimer in healthy cells (left). Electrophiles emerge during oxidative stress and alter the conformation of KEAP1 by directly adducting to the specific sensor cysteine residues (Cys) in KEAP1. Because this modification inactivates the interaction between KEAP1 and NRF2, NRF2 avoids degradation in cells exposed to oxidative stress. The stabilized NRF2 translocates into the nucleus, where NRF2 activates the transcription of its target genes by binding to NRF2 recognition sequences (i.e., ARE, EpRE, or CsMBE) as NRF2-sMAF heterodimers. Representative NRF2 target genes are separately listed in 4 categories (below).
NRF2-null mutant mice (Nrf2-KO) exhibit no obvious phenotype under specific pathogen-free conditions but fail to induce a set of oxidative and electrophilic stress-responsive genes [39]. In contrast, Keap1-null mice show neonatal lethality with hyperkeratosis in the esophagus due to NRF2-dependent overexpression of a set of cytokeratin-related genes [40]. The phenotype of KEAP1-null mice is completely reversed by simultaneous knockout of the gene that encodes NRF2 (Nfe2l2), indicating the existence of a specific association between KEAP1 and NRF2 in vivo [40]. An advanced gene-manipulation technology was used to establish a conditional targeting mouse line (KEAP1-CKO) and a hypomorphic mutant (KEAP1-KD) for the KEAP1 gene, and these mouse lines have demonstrated that KEAP1 strongly suppresses the expression of oxidative stress-responsive genes through the degradation of NRF2 under physiological conditions [41,42].
In addition to the canonical NRF2 degradation system by KEAP1 in the cytoplasm, β-transducin repeat-containing E3-ubiquitin ligase protein (βTrCP) is also involved in NRF2 degradation in the nucleus by directly associating with NRF2 in a manner that differs from KEAP1-NRF2 binding [43,44]. Oxidative stress conditions inhibit βTrCP-NRF2 binding by antagonization of glycogen synthase kinase 3 (GSK3α and GSK3β), a downstream signaling molecule of the PTEN-PI3K-AKT pathway [43,44]. Therefore, the PTEN-PI3K-AKT-GSK3 axis is considered another cellular response system to oxidative stress that is controlled by NRF2.
Evidence for the Beneficial Effects of NRF2 Activation in the Prevention of Kidney Disease Progression
Previous studies using genetically modified mice (e.g., Nrf2-KO, Keap1-KD, and Keap1-CKO mice) have demonstrated that NRF2 protects against kidney disease progression by reducing oxidative stress generated by disease conditions (Table 1). These studies have also shown that oxidative stress is involved in both the pathogenesis and progression of kidney diseases and that the accumulation of oxidative stress is a final common pathway among many types of kidney disease models in mice [42,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59].

Renal IRI is clinically caused by kidney transplantation and cardiovascular events and is one of the most common experimental models for kidney disease in rodents. We recently investigated the roles that NRF2 plays in preventing kidney disease progression from AKI to CKD using a unilateral IRI mouse model [60]. We found that, compared to the contralateral uninjured kidneys, injured kidneys slightly induce the expression of NRF2 target genes within 24 h after injury and accumulate ROS and electrophiles. This induction is transient, and the levels fall below baseline levels the day after injury (day 1), in agreement with previous reports that have utilized different animal models of nephropathy [61,62]. The tubules then undergo damage from oxidative stress caused by oxidative DNA adducts, including 8-hydroxydeoxyguanosine. This damage occurs primarily in the medulla until day 5 (Fig. 2). The damaged area expands to the cortex, and the kidney mass is reduced after day 5, with the infiltration of inflammatory cells and tubular cell death [60]. On day 14, most of the remaining tubules are damaged, and progressive fibrosis is observed in the interstitium (Fig. 2). These results suggest that IRI induces the progression of kidney damage from AKI to CKD within 2 weeks after injury [60].
![Fig. 2. Prevention of tubular damage progression by NRF2 activation during the early phase of IRI in mice. Unilateral IRI in mouse kidneys causes transient and low-level NRF2 activation in the injured kidney 1 day after injury (day 1) by generating oxidative stress [60]. Because the NRF2 induction is insufficient to prevent IRI-induced tubular damage progression, areas of the damaged tubules in the injured kidney spread from the medulla to the cortex until day 14, with a reduction of kidney mass (upper chart). The administration of an NRF2 activator (CDDO-imidazole) every 2 days from days 1-5 after injury dramatically protects the tubules from oxidative stress-mediated cell death by hyper-activating NRF2. The lower panels show Elastica-Masson staining of injured kidney sections from mice subjected to unilateral IRI on day 0. Strongly stained areas indicate functionally preserved tubular areas. Damage caused by IRI is not apparent on day 1 (left), while most of the tubules are damaged on day 14 in mice treated with vehicle (middle). The tubules are dramatically preserved by CDDO-imidazole administration on days 1-5 after injury (right).](https://karger.silverchair-cdn.com/karger/content_public/journal/ajn/45/6/10.1159_000475890/2/m_000475890_f02.jpeg?Expires=1716292727&Signature=nw7QCNUDxLYLft1VNYmGfrLcWHV2C70VnDB9XKY6xmiAJyWLR0ONF69Wa52PqkSYVlGz8bw4L2KVapYkyIBOd-z3Ba2zVJmnlxHp2s4KNchI064kp5Tt-SDUQHgtE96c1o5PGOYjuyZV0zkeMoP1YrpBbwb4fVWDkn-PK4SK4w-0GgIHFe~dUT7yeZEDmikVVUYRZ0muqQxRtMZDPkVYCuLdWIpBf~~wEUmapuHlNW4qyHzfrOS~l-BengZ3qugDfHPSV0l0OIefsCEataI5BtqyDdmBpRny7ZJHbxFlgfi6pNfG7MXj78SR45msDnUus0LoZs9ja-vtIaSTXeVVag__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Prevention of tubular damage progression by NRF2 activation during the early phase of IRI in mice. Unilateral IRI in mouse kidneys causes transient and low-level NRF2 activation in the injured kidney 1 day after injury (day 1) by generating oxidative stress [60]. Because the NRF2 induction is insufficient to prevent IRI-induced tubular damage progression, areas of the damaged tubules in the injured kidney spread from the medulla to the cortex until day 14, with a reduction of kidney mass (upper chart). The administration of an NRF2 activator (CDDO-imidazole) every 2 days from days 1-5 after injury dramatically protects the tubules from oxidative stress-mediated cell death by hyper-activating NRF2. The lower panels show Elastica-Masson staining of injured kidney sections from mice subjected to unilateral IRI on day 0. Strongly stained areas indicate functionally preserved tubular areas. Damage caused by IRI is not apparent on day 1 (left), while most of the tubules are damaged on day 14 in mice treated with vehicle (middle). The tubules are dramatically preserved by CDDO-imidazole administration on days 1-5 after injury (right).
Prevention of tubular damage progression by NRF2 activation during the early phase of IRI in mice. Unilateral IRI in mouse kidneys causes transient and low-level NRF2 activation in the injured kidney 1 day after injury (day 1) by generating oxidative stress [60]. Because the NRF2 induction is insufficient to prevent IRI-induced tubular damage progression, areas of the damaged tubules in the injured kidney spread from the medulla to the cortex until day 14, with a reduction of kidney mass (upper chart). The administration of an NRF2 activator (CDDO-imidazole) every 2 days from days 1-5 after injury dramatically protects the tubules from oxidative stress-mediated cell death by hyper-activating NRF2. The lower panels show Elastica-Masson staining of injured kidney sections from mice subjected to unilateral IRI on day 0. Strongly stained areas indicate functionally preserved tubular areas. Damage caused by IRI is not apparent on day 1 (left), while most of the tubules are damaged on day 14 in mice treated with vehicle (middle). The tubules are dramatically preserved by CDDO-imidazole administration on days 1-5 after injury (right).
The genetic loss of NRF2 worsens IRI-induced kidney damage in mice throughout the 2-week experimental period, with no or low-level induction of oxidative stress-responsive gene expression on day 1. In addition, severe infiltration of inflammatory cells and enhanced apoptosis of tubular cells are observed in injured kidneys from Nrf2-KO mice in comparison to kidneys from wild-type mice [60]. In contrast, Keap1-KD mice are resistant to the progression of IRI-induced kidney damage due to the abundant production of NADPH and glutathione, which are both essential metabolites for maintaining cellular redox homeostasis, in the injured renal cortex through the systemic activation of NRF2.
To identify the cells responsible for renal protection from oxidative damage, Keap1-CKO mice with tissue-specific expression of Cre recombinase transgenes were analyzed. Keap1-CKO mice expressing Cre in the renal epithelium of developing embryos exhibit hydronephrosis during the neonatal stage, indicating that the tubular KEAP1-NRF2 system plays a critical role in kidney morphogenesis [63,64]. Adult-onset tubule-specific Keap1 targeting, which is achieved by using the doxycycline-inducible expression system of the Cre transgene to avoid hydronephrosis, dramatically prevents the progression of IRI-induced tubular cell dysfunction, as well as that seen in the systemic reduction of Keap1 expression (Table 1) [60,65]. Cells related to inflammation accumulate in the interstitium of injured kidneys soon after IRI. T lymphocyte-specific deletion of the Keap1 gene ameliorates kidney damage [58], while myeloid cell-specific knockout of this gene shows no significant effects (Table 1) [60]. Thus, constitutive hyperactivation of NRF2 in tubules or T cells prevents AKI-to-CKD transition by enhancing cellular protection against oxidative stress.
In addition to such genetic approaches, the role of NRF2 in preventing kidney disease progression has been demonstrated in rodents via pharmacological approaches using the NRF2 activator CDDO-imidazole. Mice orally administered CDDO-imidazole on days 1, 3, and 5 after unilateral IRI treatment show mild kidney damage on day 14 compared to vehicle-treated mice (Fig. 2). Notably, CDDO-imidazole administration on days 7-13 is ineffective for kidney protection. These results indicate that enhancing NRF2 activity during the early phases of kidney disease is critical for preventing the progression of kidney damage. These results are comparable with previous studies showing that pretreatment with KEAP1 inhibitors attenuates oxidative stress and protects cells from damage in various disease models using rodents [66,67,68].
It has been reported that another KEAP1 inhibitor, CDDO-9,11-dihydro-trifluoroethyl amide (CDDO-dhTFEA, also known as dh404), worsens kidney fibrosis at high doses in the rat CKD model through activating nuclear factor κB (NFκB)-mediated inflammatory responses [69]. In fact, biochemical analyses have demonstrated that KEAP1 inhibits not only NRF2 but also NFκB, a master transcriptional activator of inflammatory cytokines [70,71]. As low-dose CDDO-dhTFEA ameliorates kidney fibrosis through the NRF2 activation without NFκB induction, NRF2 is thought to be the major target of KEAP1 [69]. However, we must consider that KEAP1 inhibitors may affect the activities of KEAP1 targets other than NRF2.
Clinical Trials of KEAP1 Inhibitors for Kidney Disease Treatment
CDDO-methyl ester, a synthetic triterpenoid based on natural products, activates NRF2 through inhibiting ubiquitination activity of KEAP1 [72]. Because CDDO-methyl ester has been considered an anti-tumor agent, a phase-1 clinical trial of CDDO-methyl ester was conducted for treating solid cancer and hematological malignancy [73]. In the trial, a 26% increase in the estimated glomerular filtration rate (eGFR) was observed in patients who were administered CDDO-methyl ester (Table 2). Therefore, CDDO-methyl ester has been featured as a promising novel drug for treating patients with renal diseases.
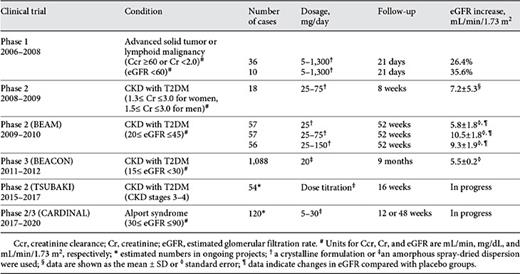
A phase-2 clinical trial (referred to as the BEAM study) was designed to verify the positive effects of CDDO-methyl ester on renal function in 227 patients with type-2 diabetes mellitus (T2DM) and stages 3-4 CKD (Table 2) [74]. The study demonstrated that patients treated with CDDO-methyl ester experience an increase in eGFR during the first 4 weeks of administration, and durable improvement for at least one year. Importantly, the improvement of eGFR was sustained at 4 weeks after stopping CDDO-methyl ester administration. It is now thought that CDDO-methyl ester improves the eGFR of kidney disease patients because of the restoration of endothelial function and/or vasodilation, followed by the preservation of the filtration bed area in the glomerulus [75,76,77]. Additionally, the inhibition of glomerular contraction and activation of glomerular filtration may occur. Although the phase-2 clinical trial confirmed quite positive effects of NRF2 activation in the prevention of kidney disease progression, the trial found that CDDO-methyl ester increased albuminuria, which is generally correlated with the risk of cardiovascular diseases in CKD patients [78,79]. Cynomolgus monkeys that were administered CDDO-methyl ester also showed a marked increase in the urine albumin-to-creatinine ratio, with decreasing expression of megalin, which is a critical regulator of urinary-albumin reabsorption in the proximal tubules, while levels of blood urea nitrogen and creatinine were reduced without histological abnormality in the kidneys [80].
Based on the positive results of the BEAM study, a phase-3 randomized clinical trial (referred to as the BEACON study) was conducted. The study included 2,185 stage 4 CKD and T2DM patients, and the increases in eGFR were consistent with those obtained in the BEAM study (Table 2) [81,82]. However, the BEACON study was terminated in 2012 due to a high incidence of cardiovascular events [81]. In this study, blood pressure and heart rate were higher in the CDDO-methyl ester group compared to the placebo group. Moreover, significant increases in the urinary albumin-to-creatinine ratio and B-type natriuretic peptide (BNP), as well as significant reductions of serum albumin and hemoglobin, were observed in patients treated with CDDO-methyl ester. Because decreases in urinary volume and urinary sodium excretion were observed in stage-4 CKD patients who were administered CDDO-methyl ester for 4 weeks [83], many of the heart failure cases appeared related to early-onset fluid overload. In addition, the severity of kidney disease may have been linked to the incidence of cardiovascular events in CDDO-methyl ester-treated patients because CKD in the BEACON study was more progressive than that in the BEAM study, in which no imbalance in cardiovascular events was observed between CDDO-methyl ester and the placebo group. Pharmacokinetics is altered by the severity of diseases, and different chemical preparations of CDDO-methyl ester have been used in these studies. Therefore, the differences in the local bioavailability of CDDO-methyl ester may be related to the different outcomes among these studies.
Ongoing Clinical Trials of NRF2 Inducers
Recently, a regression tree analysis of results from the BEACON study demonstrated that a baseline BNP (>200 pg/mL) and a history of heart failure hospitalization are closely related to the incidence of fluid overload events in the BEACON study [84]. By post hoc omitting patients with both of these predictors, the incidence of heart failure in the CDDO-methyl ester group becomes comparable to that of the placebo group in the BEACON study. Because the history of hospitalization for heart failure is similar between both groups in the BEACON study [80], an increased BNP is probably critical for the incidence of fluid overload events in CDDO-methyl ester-treated patients. CDDO-methyl ester does not change blood pressure values in healthy controls, and an 8-week administration of CDDO-methyl ester in stage 3b CKD patients does not reduce urinary volume or urinary sodium excretion [82], suggesting that patients with poor renal function may be susceptible to the vasopressor effect caused by CDDO-methyl ester.
Based on the results from the regression tree analysis of the BEACON study, a phase-2 randomized control study is currently underway to prove the renal protective effects of CDDO-methyl ester in patients with T2DM and stages 3-4 CKD without those risk factors (Table 2, the TSUBAKI study, https://clinicaltrials.gov/ct2/show/NCT02316821). The interim results of the TSUBAKI study show that inulin clearance, which has been employed as an accurate measurement of GFR, is improved by CDDO-methyl ester treatment. In addition, a phase-2 or phase-3 clinical trial of CDDO-methyl ester has been initiated in patients with Alport syndrome (the CARDINAL study; Table 2), which is a hereditary progressive renal disease that is associated with deafness and ocular diseases derived from type IV collagen abnormalities.
Future Perspectives
Although renal function is dramatically improved by NRF2 activation, the BEACON trial was interrupted due to the high incidence of cardiovascular events. By improving the treatment strategy of CKD patients without risk factors for heart failure and fluid overload, a phase-2 trial has been resumed and it is found to be yielding positive results. Therefore, CDDO-methyl ester is expected to be a first-in-class blockbuster drug for the treatment of CKD. Our study using mouse models showed that the kidneys are protected from oxidative stress-mediated damage by NRF2 activation shortly after IRI (Fig. 2) [60]. This result suggests that NRF2 activators would be effective not only in patients with developed CKD but also in patients with IRI-induced AKI, including patients undergoing cardiovascular surgery and renal transplantation [85,86].
Acknowledgments
We thank Dr. Hironori Kanda (Kyowa Hakko Kirin Co., Ltd.) for giving us professional comments on the clinical trials. This work was supported partially by Grants-in-Aid from MEXT/JSPS KAKENHI (15H04691 for N.S. and 26111002 for M.Y.) and SUZUKEN Memorial Foundation (N.S.).
Disclosure Statement
The authors have no conflicts of interest to declare.
