

Abstract
Hypohidrotic or anhidrotic ectodermal dysplasia (HED/EDA) is characterized by impaired development of the hair, teeth, or sweat glands. HED/EDA is inherited in an X-linked, autosomal dominant, or autosomal recessive pattern and caused by the pathogenic variants in 4 genes: EDA, EDAR, EDARADD, and WNT10A. The aim of the present study was to perform molecular screening of these 4 genes in a cohort of Turkish individuals diagnosed with HED/EDA. We screened for pathogenic variants of WNT10A, EDA, EDAR, and EDARADD through Sanger sequencing. We further assessed the clinical profiles of the affected individuals in order to establish phenotype-genotype correlation. In 17 (63%) out of 27 families, 17 pathogenic variants, 8 being novel, were detected in the 4 well-known ectodermal dysplasia genes. EDAR and EDA variants were identified in 6 families each, WNT10A variants in 4, and an EDARADD variant in 1, accounting for 35.3, 35.3, 23.5, and 5.9% of mutation-positive families, respectively. The low mutation detection rate of the cohort and the number of the EDAR pathogenic variants being as high as the EDA ones were the most noteworthy findings which could be attributed to the high consanguinity rate.
Ectodermal dysplasias (EDs) belong to a large clinically and genetically heterogeneous and growing group of disorders characterized by the impaired development of 2 or more ectoderm-derived structures, involving hair, teeth, nails, or exocrine glands [Freire-Maia, 1977; Lamartine, 2003; Bohring et al., 2009; Visinoni et al., 2009]. Hypohidrotic or anhidrotic ectodermal dysplasia (HED/EDA), the most frequent form of ED, is characterized by a clinical triad comprising abnormalities of the hair (hypotrichosis), teeth (anodontia, hypodontia/oligodontia, misshapen teeth), and sweat glands (anhidrosis or hypohidrosis) [Cluzeau et al., 2011]. Dysmorphic features (frontal bossing, periorbital wrinkling and hyperpigmentation, depressed nasal bridge, and prominent lips) and mammary agenesis may also be associated with HED/EDA [Cluzeau et al., 2011; Svendsen et al., 2014].
HED is predominantly inherited in an X-linked pattern (XLHED; OMIM 305100) [Chassaing et al., 2010; Cluzeau et al., 2011]. XLHED is due to mutations in the EDA gene (ectodysplasin-A), which maps to Xq12q13.1 and encodes ectodysplasin (EDA; OMIM 300451), a member of the tumor necrosis factor (TNF) family [Bayés et al., 1998]. Autosomal dominant (AD; OMIM 129490) and autosomal recessive (AR; OMIM 224900) forms of HED/EDA are encountered less frequently and result from mutations in the EDAR or in the EDARADD genes [van der Hout et al., 2008; Plaisancié et al., 2013]. Moreover, recently, it has been shown that mutations in the WNT10Agene are responsible for a broad spectrum of autosomal recessive ED [Cluzeau et al., 2011]. EDAR, encoding the EDA receptor, maps to 2q11q13 (OMIM 604095), and EDARADD,encoding the EDAR-associated death domain gene, maps to 1q42q43 (OMIM 606603). As EDA, both of the proteins encoded by these genes belong to the TNF-like EDA signaling pathway [Headon et al., 2001; Cui and Schlessinger, 2006]. Pathogenic variants in these 3 genes (EDA, EDAR,and EDARADD) result in indistinguishable phenotypes since the involved gene products all participate in the EDA-NF-κB signaling pathway, which is necessary for the initiation, formation, and differentiation of the skin appendages [Cluzeau et al., 2011; Svendsen et al., 2014]. In contrast, mutations in the WNT10Agene (OMIM 606268), encoding a protein that participates in the WNT/β-catenin/Lef-1 signaling pathway, have been shown to cause a highly variable phenotype, ranging from isolated tooth agenesis to ED syndromes such as odonto-onycho-dermal dysplasia (OODD; OMIM 257980) and Schöpf-Schulz-Passarge syndrome (SSPS; OMIM 224750) [Adaimy et al., 2007; Bohring et al., 2009; Cluzeau et al., 2011; Kantaputra et al., 2014]. The classic Wnt/β-catenin signaling pathway has been shown to play an essential role in odontogenesis, ectodermal development, and epidermal homeostasis [Tardieu et al., 2017].
The aim of the present study was to perform molecular screening to identify pathogenic variants in the EDA, EDAR, EDARADD, and WNT10A genes in a cohort of clinically diagnosed HED/EDA Turkish patients.
Patients and Methods
Patients
A total of 33 patients from 27 families with a clinical diagnosis of HED/EDA were included in the study. All patients were evaluated by expert clinical geneticists in the Medical Genetics Department, Istanbul Medical Faculty, Istanbul University. At least 3 generation pedigrees were constructed to define possible mode of inheritance, and further individuals were invited for clinical and dental evaluation.
Mutation Analysis
Genomic DNA was isolated from peripheral blood of the index patients and family members using the MagNA Pure Compact Nucleic Acid Isolation Kit (Roche, Basel, Switzerland). Primers were designed to cover all coding exons and exon-intron boundaries of EDA (NM_001399.4), EDAR (NM_022336.3), EDARADD (NM_ 145861.2), and WNT10A (NM_025216.2). Sequencing primers are available from the authors upon request. Bidirectional DNA sequencing was performed in the DNA from index patients using the Big Dye Terminator Cycle Sequencing Ready Reaction Kit version 3.1 (Applied Biosystems, Bedford, MA, USA). Identified variants were then screened in additional family members for segregation validation. One hundred control chromosomes from a healthy Turkish population were checked for the presence of all novel variants identified in this study. Population genetic data from the 1000 Genomes (http://grch37.ensembl.org/index.html), Database of Single Nucleotide Polymorphisms (dbSNP; https://www.ncbi.nlm.nih.gov/snp), and ExAC (http://exac.broadinstitute.org/) databases were also used as controls to rule out population polymorphisms. Domain information for EDA, EDAR, EDARADD, and WNT10A was retrieved from Uniprot (http://www.uniprot.org/). In silico analyses to predict the pathogenicity of the novel variants were performed using the PolyPhen-2 [Adzhubei et al., 2010] and MutationTaster2 [Schwarz et al., 2014] programs.
Results
Thirteen out of the 27 families were consanguineous. Pedigree analysis suggested probable autosomal recessive inheritance in 17 families, probable X-linked recessive inheritance in 8 families, and probable autosomal recessive or X-linked recessive inheritance in 2 families. No variants were found in 10 families, although phenotypic features of the affected patients are typical for ED (online suppl. Table 1; for all suppl. material, see www.karger.com/doi/10.1159/000499325). One patient who had a cleft lip and palate was clinically diagnosed with cleft lip/palate-ectodermal dysplasia syndrome (CLPED; OMIM 225060), but no mutation was detected in the related PVRL1 gene. Whole exome sequencing is planned for these 10 families to elucidate the molecular basis of the disease. Novel genes may be associated with their etiology.
In 17 (63%) of the 27 families, variants were detected in at least 1 of the 4 known ED genes. Mutations and phenotypic features of the mutation-positive families are summarized in Table 1. The EDARand EDA pathogenic variants were identified in 6 families each, the WNT10A in 4, and the EDARADD in 1, accounting for 35.3, 35.3, 23.5, and 5.9% mutation-positive families, respectively. The pedigrees of the mutation-positive families are shown in online supplementary Figure1.
Phenotype and genotype of HED/EDA cohort
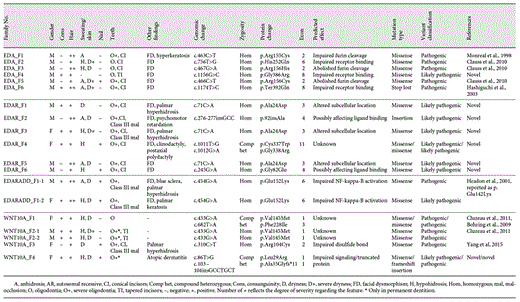
EDAR
Five novel EDAR mutations were identified in 6 index patients from 6 families and the variants segregated consistent with an autosomal recessive inheritance. In 5 families, mutations were at homozygous state and in 1 at compound heterozygote state. Three families were consanguineous. The remaining 3 families were apparently not consanguineous, but the parents of each index patient originated from the same villages, suggesting possible consanguinity. The c.276-277insGCC change was predicted to be disease-causing by MutationTaster. The remaining alterations in the EDAR gene were missense variants predicted to be possibly or probably damaging as well as disease-causing according to in silico analyses. The c.71C>A (p.Ala24Asp) variant, detected in 3 index patients, resides in exon 3 and is predicted to affect the signal peptide region of the corresponding protein. The c.276-277insGCC and c.245G>A variants are located in an extracellular cysteine-rich domain and are assumed to alter ligand binding [Zonana and Huttner, 2016]. The c.1011T>G and c.1012G>A variants in exon 11 detected in the EDAR_F4 proband correspond to the intracellular domain of the protein and were found to be in the transposition according to segregation analysis (Fig. 1) confirming compound heterozygosity.

Domains of ED-related genes (WNT10A, EDA, EDAR, and EDARADD) and the mutational distribution. Novel mutations identified in this study are in bold print.
Domains of ED-related genes (WNT10A, EDA, EDAR, and EDARADD) and the mutational distribution. Novel mutations identified in this study are in bold print.
All patients carrying EDAR variants presented with hypo/anhidrosis, sparse hair, and facial dysmorphism to varying degrees, comprising periorbital wrinkling and hyperpigmentation. They also exhibited missing primary and permanent teeth and conical permanent incisors. The mean number of congenitally missing permanent teeth was 21 (range 17-26). Parents carrying these variants in the heterozygous state were clinically evaluated and found to exhibit no ectodermal phenotype.
EDA
A total of 6 variants were identified in EDA: 5 missense variants and 1 variant leading to the loss of a stop codon. One missense variant, c.1156G>C (p.Gly387Arg), was novel and predicted to be probably damaging and disease-causing by in silico analyses. c.1156G>C resides in the TNF-like domain of the gene and is assumed to impair receptor binding (Fig. 1). The c.463C>T [Monreal et al., 1998], c.756T>G [Clauss et al., 2010], c.467G>A [Clauss et al., 2010], c.466C>T [Clauss et al., 2010], and c.1174T>C [Yang et al., 2015] variants identified in the remaining 5 patients had been reported previously.
All EDA mutation-positive affected males had the classic XLHED phenotype, including severe hair, sweat gland, and teeth anomalies (Fig. 2). Three of the 6 obligate carrier females displayed mild teeth and hair anomalies, and 3 had no symptoms. The mean number of permanent congenitally missing teeth in affected males was 21.6 (range 12-27).
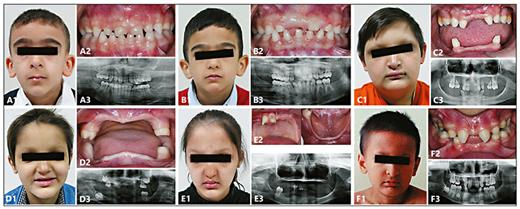
Clinical photographs and radiographs. Frontal facial view (1), intraoral view (2), and panoramic radiograph (3). A, B WNT10A_F2 siblings showing normal primary dentition and severely affected permanent dentition. C EDA_F3 proband showing facial dysmorphism and severe oligodontia. D, E EDARADD_F1 siblings showing severe facial dysmorphism and severe oligodontia. F EDAR_F5 proband showing moderate oligodontia.
Clinical photographs and radiographs. Frontal facial view (1), intraoral view (2), and panoramic radiograph (3). A, B WNT10A_F2 siblings showing normal primary dentition and severely affected permanent dentition. C EDA_F3 proband showing facial dysmorphism and severe oligodontia. D, E EDARADD_F1 siblings showing severe facial dysmorphism and severe oligodontia. F EDAR_F5 proband showing moderate oligodontia.
WNT10A
A total of 5 different pathogenic variants in WNT10A, 2 being novel, were identified in 5 patients from 4 families. Three known variants (c.433G>A, c.682T>A, and c.310C>T) were missense [Bohring et al., 2009; Cluzeau et al., 2011; Yang et al., 2015]. The WNT10A_F4 patient showed compound heterozygosity for 2 variants, 1 missense (c.86T>G), and 1 frameshift insertion (c.103-104insGCCTGCT). The c.86T>G variant is located in exon 1, which encodes for the signal peptide, and the c.103-104insGCCTGCT change in exon 1 is assumed to encode a truncated protein (Fig. 1). Both variants were predicted to be disease-causing and/or possibly damaging by in silico analyses.
Patients with WNT10A pathogenic or possibly pathogenic variants did not have facial features such as periorbital wrinkling or hyperpigmentation (Fig. 2). Hair sparsity was milder when compared with the patients who have mutations in other ED/HED genes, while the severity of hypo/anhidrosis did not differ. The mean number of permanent congenitally missing teeth was 19.2, with permanent tooth agenesis being more severe than primary tooth agenesis.
EDARADD
Sequencing of the EDARADDgene led to the identification of only 1 previously reported variant [Headon et al., 2001] in 2 siblings born to first-degree cousins. The p.Glu152Lys (NM_145861) variant was reported as p.Glu142Lys (NM_080738) by Headon et al. [2001] due to different protein isoforms being used. Both siblings exhibited severe oligodontia of the primary and permanent teeth. They also exhibited an abnormal crown morphology in the primary and permanent dentition as well as skeletal Class III malocclusion due to maxillary hypoplasia. The siblings showed severe facial dysmorphism, sparse hair, dry skin, and spoon-shaped nails. The male sibling had anhidrosis, and the affected female sibling had hypohidrosis (Fig. 2).
Discussion
Twenty-seven families clinically diagnosed with HED/EDA were included in the cohort. Four known ED genes, EDA, EDAR, EDARADD, and WNT10A, were sequenced, and 17 pathogenic variants, 8 being novel, were identified in 63% (17/27) of the cohort. Cluzeau et al. [2011] examined all 4 ED-related genes in a large cohort of 65 unrelated patients, 61 of which presented with HED/EDA, and concluded that the 4 ED genes accounted for 92% of their cases. In a recent cohort of 133 Italian ED individuals, pathogenic changes were identified in 106 subjects (80%). EDA, WNT10A, EDAR, and EDARADD variants were detected as 45, 25, 7.5, and 1.5%, respectively [Guazzarotti et al., 2015]. In another large cohort study, 124 European HED patients were investigated by Sanger sequencing and MLPA analyses of EDA, EDAR, EDARADD, TRAF6, and EDA2R, revealing pathogenic changes in 101 out of 124 HED individuals (81.5%). Patients with pathogenic variants in the EDA, the EDAR, and the EDARADD genes constituted 88, 9, and 3% of the cases, respectively [Wohlfart et al., 2016a]. Compared to these previous large cohorts, the cohort reported herein exhibited a lower variant detection rate that can partially be explained by the limitations of Sanger sequencing, which cannot detect exonic deletions, duplications, or regulatory sequence variants. Furthermore, given the high rate of consanguinity in our cohort, the lower variant detection rate may also stem from variants in as yet unidentified novel genes with autosomal recessive inheritance patterns. In another ED cohort of 40 patients from 35 Mexican families, EDA, EDAR, and EDARADD genes were sequenced, and 16 different variants were identified only in 21 families (60%) [Salas-Alanis et al., 2015]. The WNT10Agene was not included in the study, so this may have led to the lower mutation detection rate. The vast majority of HED/EDA cases carry mutations in EDA, with a reported prevalence of 58% [Cluzeau et al., 2011] and 88% [Wohlfart et al., 2016a]. To date, more than 300 pathogenic variants in EDA have been reported (Human Gene Mutation Database) [Stenson et al., 2003]. The present study revealed a lower variant rate in EDA (35.3%) compared to those previously reported cohorts. EDAR variants in our study were detected in 35.3% of the patients, similar to the rate of EDAvariants. This is in contrast to previous studies, which reported the incidence of EDAR variants at 9% [Wohlfart et al., 2016b] and 16% [Cluzeau et al., 2011]. The high rate of EDAR variants in the present study is probably associated with the high incidence of consanguineous marriages in our cohort, which increases the likelihood of an autosomal recessive inheritance pattern.
The phenotypes associated with EDA, EDAR, and EDARADDvariants are indistinguishable, and the clinical features among these patients were typical, so that a diagnosis of ED was often possible even after a cursory examination. Ectodermal symptoms including hypo/anhidrosis, sparse hair, and oligodontia were consistently present in all cases; facial dysmorphism and dry, translucent, and velvety skin were also frequent features. In contrast, the phenotypic features of our patients carrying WNT10A variants were consistent with those reported by Cluzeau et al. [2011]. Although patients harboring WNT10A variants have a marked dental phenotype, they show no dysmorphic facial characteristics suggestive of a clinical diagnosis of HED. Bergendal et al. [2016] suggested that most WNT10A mutation-positive patients exhibited a thin upper lip as a dysmorphic feature, although in our small cohort, only 2 patients with the WNT10A variants had thin upper lips. The most significant feature of individuals carrying WNT10A changes is reported to be severe oligodontia in permanent dentition with apparently normal or comparatively less disturbed primary dentition [Bohring et al., 2009]. Our findings further support this observation. In the present study, 3 out of 5 individuals (F1, F2-1, and F2-2) carrying the WNT10A variants had a full set of deciduous dentition, whereas permanent dentition showed severe oligodontia. The other 2 WNT10A variant carriers exhibited permanent dentition that was more severely affected than primary dentition. Another typical dental feature of patients with the WNT10A mutations is the presence of tapered permanent incisors [Bohring et al., 2009; Bergendal et al., 2016]. In the present study, 3 patients carrying the WNT10A variants showed tapered incisors.
Conclusion
In this study, we assessed the presence of pathogenic variants in 17 HED/EDA families. Eight novel and 9 known variants were identified in the EDA, EDAR, EDARADD, and WNT10A genes, expanding the mutational spectrum of HED/EDA. The rate of EDAR variants was found to be higher than that observed in previous studies. Thus, in populations with a high rate of consanguinity, if pedigree analyses rule out an X-linked mode of inheritance, we highly suggest that EDAR be the first gene to be tested in HED/EDA. We also suggest that the WNT10Agene should be the first to be considered if the patient has severe oligodontia in permanent dentition and mildly affected or unaffected primary teeth with no facial dysmorphism.
Acknowledgment
We are grateful to the patients and their families for participating in this study.
Statement of Ethics
The study has been conducted in accordance with the Declaration of Helsinki, and ethical approval has been obtained from the local ethic committees of the Istanbul Medical Faculty at Istanbul University (2008/1194 for CRANIRARE and 2012/743-1061 for CRANIRARE-II) and the Koç University (2015. 120.IRB2.047 for CRANIRARE-II). All participants and/or their legal representatives gave written informed consent both for molecular testing and publication of accompanying photographs.
Disclosure Statement
The authors have no conflicts of interest to declare.
Funding Sources
This work was supported by the Programme Hospitalier de Recherches Cliniques (PHRC) and the Scientific and Technological Research Council of Turkey (TUBITAK), grants 108S418 and 112S398 to H.K., and the overall consortiums, CRANIRARE and CRANIRARE-2, of the European Research Area Network projects (E-RARE).
Author Contributions
A. Smahi and H. Kayserili conceived and designed the experiments. Z. Oya Uyguner, E. Bal, and S. Hadj-Rabia performed the experiments. Y. Guven, U. Altunoglu, E. Yücel, and E. Bal analyzed the data. M. Koruyucu, E. Bahar Tuna, S. Çıldır, and O. Aktoren contributed to data acquisition. Y. Guven and H. Kayserili wrote the paper. All authors gave final approval and agree to be accountable for all aspects of the work.
