

Abstract
Background: There has been huge progress over the last 30 years in identifying the familial component of breast cancer. Summary: Currently around 20% is explained by the high-risk genes BRCA1 and BRCA2, a further 2% by other high-penetrance genes, and around 5% by the moderate risk genes ATM and CHEK2. In contrast, the more than 300 low-penetrance single-nucleotide polymorphisms (SNP) now account for around 28% and they are predicted to account for most of the remaining 45% yet to be found. Even for high-risk genes which confer a 40–90% risk of breast cancer, these SNP can substantially affect the level of breast cancer risk. Indeed, the strength of family history and hormonal and reproductive factors is very important in assessing risk even for a BRCA carrier. The risks of contralateral breast cancer are also affected by SNP as well as by the presence of high or moderate risk genes. Genetic testing using gene panels is now commonplace. Key-Messages: There is a need for a more parsimonious approach to panels only testing those genes with a definite 2-fold increased risk and only testing those genes with challenging management implications, such as CDH1 and TP53, when there is strong clinical indication to do so. Testing of SNP alongside genes is likely to provide a more accurate risk assessment.
Introduction
The inherited aggregation of breast cancer has been intimated for over 130 years [1]. A family history of breast cancer in a first-degree relative has been linked to an approximate 2-fold relative risk [2]. Approximately 4–5% of all breast cancers are thought to result from inheriting high-risk dominantly inherited pathogenic variants (PV) [2] (but only 2–3% result from BRCA1/2 mutations). However, around 27% are thought to have some form of inherited component, as reported in twin studies [3]. This means that the majority of the inherited components of breast cancer are likely due to polygenic inheritance rather than inheritance of a single gene PV.
Relatively few women with breast cancer present with a clear pattern of cancers in the family consistent with that reported by Broca [1]. However, clusters of breast cancers within families, particularly occurring at younger ages, are not infrequent and account for approximately 5% of cases overall [3]. As even genes conferring the highest risk do not cause breast cancer in every woman in their lifetime unaffected carriers of the “cancer gene” may mask true inherited families and chance clusters may mimic inherited disease.
In many instances of familial breast cancer, there is a high incidence of other tumors, notably ovarian, prostate, and pancreatic cancer or in rarer instances sarcomas and brain tumors. Empiric risks for women who have particular types of family history have been calculated [2] and have led to criteria for proposed enrolment in familial breast cancer clinics. However, increasingly risk is informed by undertaking panel tests of genes linked to breast cancer risk and also tests for multiple common variants in a polygenic risk score (PRS).
This paper will cover all of the known genes and genetic variants linked to breast cancer risk from the discovery of the first bona fide breast cancer genes including BRCA1 through to the development of PRS from multiple single-nucleotide polymorphisms (SNP) discovered through genome-wide association studies.
Generally, genes and SNP have been divided into high-risk genes with an OR ≥4.0 (lifetime risks in European populations ≥40%), moderate risk genes with an OR ≥2.0 but < 4.0 (lifetime risks in European populations ≥20% but < 40%), and genetic modifiers with an OR of 1.01–1.5 (Fig. 1).

Relative risk and allele frequencies of high-, moderate-, and low-risk genetic variants.
Relative risk and allele frequencies of high-, moderate-, and low-risk genetic variants.
Molecular Genetics
High-Risk Breast Cancer Genes
BRCA1
Molecular geneticists started to try to identify breast cancer genes in the mid 1980s. By 1990 the first gene (BRCA1) had been identified on chromosome 17q by linkage analysis in breast cancer families [4]. It quickly became apparent that the locus later termed BRCA1 conferred a risk of both breast cancer and ovarian cancer [5], as originally predicted by Henry Lynch and Krush [6] some 20 years earlier. The gene itself was cloned in 1994 [7]. The BRCA1 gene is very large, with a 7,207-bp transcript. It has only limited homology with any previously identified human sequence, and its function is not fully clarified. The main functions are homologous repair of double-stranded DNA breaks and transcriptional activation. BRCA1 is predominantly a breast/ovarian cancer gene conferring lifetime risks of 50–85% for breast cancer and 30–60% for ovarian cancer [8-11]. Initial assessments based on high-risk families tended to provide risk estimates only appropriate for that setting [8, 9]. However, attempts to strip out the biases of familial ascertainment providing very low estimates were not really realistic in relation to the real situation outside of those identified with no family history of breast or ovarian cancer [12]. The best current estimates are based on prospective studies [10, 11], which lack the ascertainment bias of the earlier historic studies. Nonetheless, clinicians and genetic counsellors should refrain from providing a very specific risk (e.g., 72% breast cancer risk by age 80 years for BRCA1) [11] as the risks will vary with nongenetic risk factors such as reproductive factors as well as the degree of family history. These factors can be incorporated into a model developed from the BOADICEA algorithm called CanRisk [13]. The addition of a PRS from SNP (see below) is likely to provide an even more accurate likelihood of the breast and ovarian cancer risk, with overall likelihoods varying from as little as 45% to > 95% for female breast cancer in BRCA1 [14]. The risks of estrogen receptor negative (ER–) breast cancer varied from 59 to 83% at the 5th and 95th percentiles, while the risks of ovarian cancer by age 80 years were 30 and 59% for BRCA1. The pathology of BRCA1-related cancers is fairly specific, with the great majority of ovarian cancers being high grade serous and around 70% of breast cancers being ductal triple negative (negative for HER2, ER, and progesterone receptor) [15]. There is no evidence of an increased risk of mucinous ovarian cancer [15, 16] and this should be discounted in algorithms to predict the likelihood of BRCA1, as it is in the pathology-adjusted scoring system [17]. Similarly, HER2 positivity is uncommon in BRCA1 PV carriers and should reduce the likelihood of finding a causative variant [17]. There is no strong convincing evidence for the risk of other cancers in BRCA1 carriers [18]. A number of other cancers have been shown to potentially be related, including early-onset prostate cancer [19, 20], colorectal cancer [19, 21], endometrial cancer [19, 22], and pancreatic cancer [20]. However, the OR for these cancers are generally < 3-fold and have not been convincingly replicated [18, 23, 24]. Although there may be a specific risk of high-grade serous endometrial cancer as this translates to < 10% of all endometrial cancers, this is unlikely to translate into a substantial OR for endometrial cancer as a whole [24]. Furthermore, a study comparing BRCA1 and BRCA2 found that it was BRCA2 that harbored the truly more heterogeneous cancer risk [25]. Indeed, the only prospective study of early detection outside of breast and ovarian cancer showed no evidence of benefit for prostate cancer in BRCA1, with cancer incidence not being significantly increased compared to controls [26]. Overall, BRCA1 carriers should be advised that there is unconvincing evidence of substantial increased risks of cancers beyond breast and ovarian cancer and that if there is any increased risk it is unlikely to be sufficient to warrant early detection measures.
BRCA2
Soon after the BRCA1 locus was identified, it became clear that many large breast cancer kindreds, particularly those with an affected male, were not accounted for by BRCA1. A second locus, i.e., BRCA2, was mapped by family linkage analysis to chromosome 13q in 1994 and within a year the gene had been isolated [27]. The size of the gene is even greater than that of BRCA1 (11,386 bp), with which it shows some homology particularly with regard to homologous repair and cancer predisposition. BRCA2 PV confer female lifetime risks of 40–87% for breast cancer and 10–30% for ovarian cancer [8-11, 28]. The effect of a breast cancer family history on breast cancer risk is even greater for women with BRCA2 PV [8-11, 28]. Estimates of the BRCA2 breast cancer risk have been as low as 38%, stripping out any additional familial risk effects [12]. However, these studies, which rely on historic data, include women born before 1930 in whom breast cancer risks are much lower than those for modern day women [28]. Again, the best current risk estimates are based on prospective studies [10, 11]. Like for BRCA1, clinicians and genetic counsellors should refrain from providing a very specific risk, such as 69% breast cancer risk by age 80 years for BRCA2 [11], as similarly to BRCA1 the risks will vary with nongenetic risk factors such as reproductive factors as well as the degree of family history. These factors can be incorporated into a model developed from the BOADICEA algorithm called CanRisk to give a personalized risk assessment based on germline genetic and other known risk factors [13]. The addition of a PRS from SNP (see below) is likely to provide an even more accurate likelihood of breast and ovarian cancer risks, with overall likelihoods varying from as little as 43% to > 95% for female breast cancer in BRCA2 [14]. The risks breast cancer varied from 57–81% at the 5th and 95th percentile, but they were as little as 43% with no family history and as high as 85% with a family history of breast cancer. Likewise, the ovarian cancer risks by age 80 years at the 5th and 95th percentile were 10 and 28% for BRCA2. The pathology of BRCA2-related breast cancer is not as specific as that for BRCA1, although there is a trend toward higher-grade ductal ER+ PR+ HER2–, with only 16% of cases being triple negative, but unlike BRCA1 the likelihood of triple-negative increases with age [15]. HER2 positivity is less common but not as infrequent as for BRCA1 PV carriers [17]. The great majority of ovarian cancers are high-grade serous [15]. Similarly to BRCA1, there is no evidence of an increased risk of mucinous ovarian cancer [15, 16]. Males have a substantially increased risk of breast cancer, with a lifetime risks of 5–14% [29, 30]. One study using a PRS found that the risk of breast cancer by age 80 years is 5% for men at the 5th percentile of the PRS and 14% for men at the 95th percentile [30]. In addition to breast and ovarian cancer risk, BRCA2 PV clearly also predispose to prostate cancer (OR = 2.5–6.3), pancreatic cancer (OR = 3.5–5.9), gastric cancer (OR = 2.4–2.59), and various skin cancers including melanoma, all of which have been validated in at least 2 studies [18, 31-33]. Though rare, ocular (uveal) melanoma appears to also be strongly linked [18, 34]. As above, a study comparing BRCA1 and BRCA2 found that BRCA2 conferred the greater cancer risk beyond breast and ovarian cancer [25]. The prospective IMPACT study of early detection of prostate cancer showed an excess risk in BRCA2 carriers and that PSA was effective at earlier detection of the predominantly more aggressive higher Gleason score prostate cancer [26].
Li-Fraumeni Syndrome and TP53
Even before the identification of BRCA1/2, the TP53 gene had been implicated in hereditary breast cancer as part of the Li-Fraumeni cancer family syndrome [35]. It was, however, recognized that this accounted for only a very small proportion of breast cancer families and subsequent studies confirmed this impression. Nonetheless, mutations in TP53 may account for almost as many breast cancers in patients ≤30 years of age as BRCA2 [36], and diagnosis at an age ≤30 years is a criterion for testing by the Chompret criteria [37]. Overall, 2–8% of breast cancers in patients aged ≤30 years harbor a TP53 germline PV and these are more common with HER2+ invasive disease and high-grade comedo-DCIS [38]. Detection rates drop dramatically after 30 years of age, and testing of women after age 45 years with no previous malignancy and no other element of Chompret criteria fulfilled (no typical Li-Fraumeni cancer in a close relative) is not recommended [37]. This is due to the much likelier possibilities of identifying clonal hematopoiesis of indeterminant potential (CHIP) or a variant of uncertain significance, which could be misclassified as likely pathogenic erroneously [37]. People identified as carrying PV in TP53 have a very high lifetime risk of malignancy, although this may vary with the exact variant, with dominant negative missense variants in the core binding domain conferring the highest risks [37]. There are particularly high risks of sarcoma, especially osteosarcoma and embryonal rhabdomyosarcoma, gliomas, and other brain malignancies such as choroid plexus carcinoma and SHH-medulloblastoma as well as adrenocortical carcinoma [37]. Although screening has been shown to have an impact, with whole-body MRI, breast MRI, and dedicated brain MRI now being recommended, the psychological impact of being identified as a TP53 PV carrier or erroneously being misdiagnosed is considerable.
Cowden Syndrome and PTEN
The PTEN gene on chromosome 10q has been identified as the causal gene in Cowden syndrome, in which early-onset breast cancer is associated with a variety of other features including hamartomas of the skin and mucous membranes, thyroid adenomas and cancer, colonic polyps (including juvenile polyps), and craniomegaly [39]. While prospective breast cancer risk data is lacking due to its rarity, women with Cowden syndrome are at a high breast cancer risk and ought be offered equivalent high-risk breast cancer risk reduction measures. Additional tumor risks include thyroid cancer and endometrial cancer. Due to its rarity and usual syndromic features of marked macrocephaly, PTEN is rarely found on gene panels in the absence of diagnostic features (< 0.1%) [40].
Peutz-Jeghers Syndrome and STK11
STK11 (LKB1) is associated with the dominantly inherited condition Peutz-Jeghers syndrome (PJS), which is characterized by typical benign PJS polyps throughout the gastrointestinal tract and muco-cutaneous pigmentation (particularly on the lips). The breast cancer risk is probably between 40 and 60% lifelong [41]. Due to the rarity and distinct clinical features of PJS, STK11 PV are extremely rarely found on gene panels (Table 1).
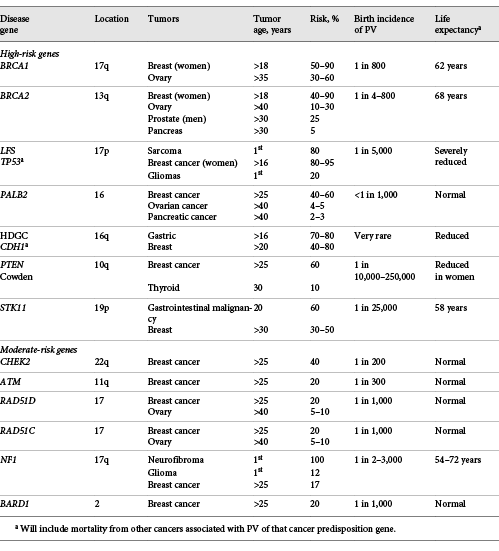
Hereditary Diffuse Gastric Cancer and CDH1
Mutations in the gene CDH1 causes the dominantly inherited condition hereditary diffuse gastric cancer. Women with mutations in CDH1 again have a 40–60% lifetime risk of breast cancer, often of lobular histology [42]. Due to its rarity and frequent family history of diffuse gastric cancer, CDH1 is rarely found on gene panels (< 0.1%) [40], and some countries recommend that it be excluded from gene panels in the absence of familial lobular cancer or diffuse gastric cancer due to the consequences of erroneously identifying women as PV carriers [43].
PALB2
Upon first identification, PALB2 was identified as a moderate-penetrance breast cancer susceptibility gene. In a case-control study, 10 truncating PV were identified in 923 individuals with familial breast cancer but no PV were identified in 1,084 healthy controls (p = 0.0004) [44]. Despite the absence in controls, the relative risk of breast cancer associated with a PV in PALB2 was only assessed as moderate in this discovery paper with a 2-fold OR [44]. However, subsequent assessment of risk in families confirmed a high risk, with an OR of 7.18 (95% CI 5.82–8.85; p = 6.5 × 10–76), and a lifetime risk of around 50% [45]. As with BRCA2 the risks vary based on the family history and presumably the SNP PRS profile [45]. There is likely a small increase in the ovarian cancer risk and the evidence for an increased risk of pancreatic cancer is also fairly robust [45].
Moderate-Risk Genes
Ataxia-Telangiectasia
Ataxia-telangiectasia (ATM) was the first moderate-penetrance breast cancer gene for which there was strong clinical evidence. The possibility that ATM could be a breast cancer susceptibility gene was first proposed nearly 40 years ago when epidemiologists suggested that relatives of patients with an autosomal recessive condition called ATM, which predisposes to cancer in childhood, particularly lymphoid cancers, had an increased risk of breast cancer. The role of ATM in breast cancer susceptibility has been investigated in many studies. The first conclusive study identified 12 mutations in 443 familial breast cancer cases and 2 in healthy controls (p = 0.0047), suggesting that the relative risk in female ATM mutation carriers is 2.37 [46]. A 2- to 3-fold OR for ATM has been confirmed in many studies, consistent with lifetime risks of 20–30% [47]. There is nonetheless evidence of one dominant negative missense variant in ATM c.7271T>G, i.e., p.(Val2424Gly), that is consistent with a 60% lifetime risk [48, 49]. However, the risks for other missense variants may be < 2-fold and these are difficult to classify [47, 50].
CHEK2
CHEK2 is a gene that encodes a cell cycle checkpoint protein kinase that phosphorylates TP53 and BRCA1 and is involved in DNA repair. The relative risk of breast cancer in carriers of the CHEK2 c.1100delC allele was estimated to be 2.2 [51], and it appears to be similar for other truncating PV in CHEK2 [47]. Missense variants can be associated with increased risks, but these are generally below 2-fold [47]. PRS appear to be helpful in more accurately defining risk in CHEK2 PV carriers [52], and individual risks can be assessed taking into account reproductive and other risk factors as for BRCA1/2 [13]. There are no clear additional risks to CHEK2 PV carriers, although increased risks of colorectal and prostate cancer have been found in some studies.
Neurofibromatosis 1
Women with the inherited tumor-prone condition neurofibromatosis 1 (NF1) are now thought to be at a moderately increased risk of developing breast cancer [53-55]. There is a particularly high OR in patients aged < 50 years, with a 10% risk by that age, after which the risk levels off [54]. The breast tumors associated with NF1 PV have adverse pathological features, with higher proportions of grade 3 ER– and HER2+ and poor survival [55, 56]. As NF1 is an easy syndromic diagnosis with clinically easily detectable features (café au lait patches, skin neurofibromas) in the vast majority of individuals gene panel testing is unlikely to be required and it is very unlikely that testing will find a PV in NF1 without these features [57].
Each of these moderate penetrance genes makes a relatively small contribution to the overall familial risk of breast cancer. Compared to the 16–18% of familial risk accounted for by mutations in BRCA1 and BRCA2, currently it is estimated that moderate-penetrance breast cancer susceptibility genes only account for 6–7% of the familial risk (Fig. 2). In keeping with findings in BRCA1 and BRCA2, most of the PV in these genes lead to premature protein truncation through nonsense codons or translational frameshifts. A very small number are possibly due to missense sequence variants. The moderate-penetrance breast cancer susceptibility genes each harbor multiple different rare PV.

Proportion of the familial component caused by known genetic factors. GWAS, genome-wide association study.
Proportion of the familial component caused by known genetic factors. GWAS, genome-wide association study.
Other Probable Moderate-Risk Genes
Three further genes that had previously been linked to breast cancer risk, but for which validation studies were inconclusive, were also supported in the BRIDGES study of 60,000 cases and 53,000 controls as having close to a 2-fold relative risk. These are the ovarian cancer genes RAD51C (OR = 1.93; 95% CI 1.20–3.11) and RAD51D (OR = 1.80; 95% CI 1.11–2.93) as well as BARD1 (OR = 2.09; 95% CI 1.35–3.23). All 3 genes were particularly strongly linked to triple-negative breast cancer [47].
Genes Probably Spuriously Linked to Breast Cancer
BRIP1
In 2006 BRIP1, a BRCA1-interacting helicase (also known as BACH1), was also identified as a probable rare moderate-penetrance breast cancer susceptibility allele. In a case-control study, 9 mutations were identified in 1,212 familial breast cancer cases compared to 2 in 2,081 healthy controls (p = 0.003); the relative risk of breast cancer in monoallelic carriers of BRIP1 mutations is 2.0 [58]. More recent work has, however, shown that the original link with breast cancer was spurious [59], and a large case-control study of over 60,000 cases showed an OR of only 1.11 with a 95% CI (i.e., 0.80–1.53) excluding a 2-fold risk [47]. At present, germline BRIP1 mutations are considered a risk factor for postmenopausal ovarian cancer.
In addition to BRIP1 a number of other genes identified as significantly linked to breast cancer have been shown by the BRIDGES study to be likely spurious [47]. These include NBN (OR = 0.90; 95% CI 0.67–1.20), FANCM (OR = 1.06; 95% CI 0.90–1.26), RECQL (OR = 0.84; 95% CI 0.64–1.10), RAD50 (OR = 1.08; 95% CI 0.83–1.40), and XRCC2 (OR = 0.96; 95% CI 0.47–1.93).
Missing Heritability of Breast Cancer Predisposition
While recent population-based studies have estimated the frequency of germline BRCA1/2 PV to be as high as 1 in 200 [47, 60], outside of strong founder populations such as the Icelandic and Jewish, this is insufficient to account for more than 20% of the heritable component of breast cancer and only about 2% of all breast cancers [61, 62]. The discrepancy has become even greater with the recognition that many women with germline mutations in either BRCA1 or BRCA2 have an average lifetime risk of developing breast cancer (“penetrance”) of around 65–70% or less rather than the 85–90% originally generated from high-risk families. While there may be families and individuals with hitherto undetected germline BRCA1/2 mutations, or novel mechanisms of disruption, e.g., epigenetic silencing [63], structural variants, or deep intronic variants causing splicing that are missed by standard DNA testing, the evidence for these reducing sensitivity by more than 2–5% is limited [64]. It is nonetheless likely that the majority of the missing heritability is due to the presence of undiscovered low-penetrance genetic modifiers (Fig. 2).
Low-Risk Genetic Susceptibility
For longer than a decade genome-wide association studies have been performed in order to identify associations between common variants and disease. This has led to the robust identification of more than 300 SNP that are associated with breast cancer risk [65-83]. These common low-risk alleles only confer a small risk by themselves, but when combined in a PRS the SNP provide a more informative risk estimate. It has been estimated that these SNP currently explain 28% of the familial risk of breast cancer [83] (Fig. 2), though much of the remaining 45% of the familial component of breast cancer yet to be discovered is thought to be likely more SNPs.
Due to the increase in the number of SNP found to be associated with breast cancer, and the increasing number of patients included in these association studies, subgroup analysis has shown that there are several SNP that are more strongly associated with ER-negative disease than with ER-positive disease and vice versa [75, 76, 79, 83-87]. Similarly, it has been proposed that these SNP potentially can be utilized to modify the risk of PV carriers (see above) [88].
Numerous studies have validated the predictive power of these SNP, as well as their added value for existing prediction models based on classical risk factors [88-94].
The majority of the SNP related to breast cancer risk have been found and validated using data of studies participating in the Breast Cancer Association Consortium (BCAC), which is a collaboration involving over 100 international case control studies. The vast majority of studies included in the BCAC are performed in populations of European ancestry. Therefore, the current PRS is mostly applicable to women of European ancestry. With some adjustments, the current PRS may be suitable for women of Asian ancestry [95]. However, for women with any other ancestry, the current PRS provide no correct predictive estimate of the breast cancer risk.
Application of an SNP PRS alongside a risk evaluation tool such as Tyrer-Cuzick and a measurement of mammographic density can identify about 45% of breast cancers in the top 20% of the population [94].
Detection of Mutations in Known Breast Cancer Genes
While varied laboratory techniques were used previously to detect germline mutations of the known high-risk breast cancer genes, these were time consuming, had a limited sensitivity, and were offered to families in which there was a high likelihood of detection of a germline mutation. The introduction of Sanger sequencing to detect intragenic sequence variants followed by the addition of MLPA (multiplex ligation-dependent probe amplification) to detect whole exon or gene deletions (accounting for 10–15% of germline BRCA1/2 [15–20% BRCA1 and 4–5% BRCA2] PV) has resulted in a much higher mutation detection rate. In recent years, advances in genomic technologies with massively parallel sequencing approaches have lowered the costs further and increased the sensitivity of mutation detection, further enabling testing to be offered to a wider patient population.
With these technological advances comes a separate and new set of challenges including accurate classification of the variants identified [96]. Equally important is ensuring that, for mutations detected in the mainstream setting, at-risk family members are offered testing for the familial PV where appropriate [97]. Where specific founder PV are present in certain population groups, laboratories will often retain a Sanger sequencing-based specific assay for their detection. For example, as approximately 2–2.5% of Ashkenazi Jewish women carry 1 of 3 specific mutations (BRCA1 c.68_69delAG, BRCA1 c.5266dupC, or BRCA2 c.5964delT), which collectively account for around 60% of all familial breast cancers in this population group, testing for these alone will provide meaningful predictive information even when negative. For example, exclusion of the 3 PV in an Ashkenazi Jewish woman who has a family history of breast cancer will reduce her lifetime risk by 40–50%, as the 3 mutations make up 50% of the inherited risk. A similar calculation applies to Icelandic women, among whom the BRCA2 c.771_775del p.(Asn257LysfsTer17) founder mutation accounts for a high proportion of breast cancer families [98]. Given the relatively low cost of full BRCA testing and of undertaking panels, testing for common PV is now much less commonly carried out unless these account for the great majority of hereditary high-penetrance variants in a country or population.
Breast Cancer Panels
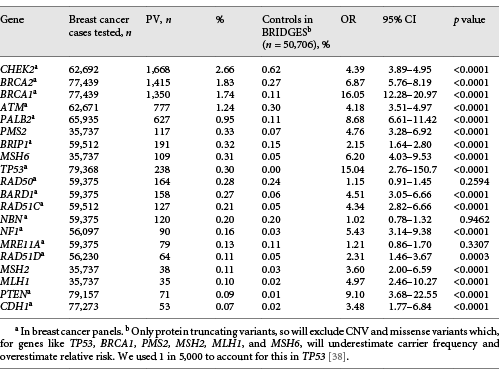
Until around 2014 the great majority of breast cancer genetic testing was bespoke testing and suggested that NBN did qualify based on 1 protein-truncating variant, i.e., c.657del5, but BRIDGES did not confirm this for protein-truncating variants as a whole [47]. An idea of detection rates from commercial testing can be gained from testing of breast cancer cases with BRCA1/2 alone, with targeted testing of TP53 and syndromic genes such as PTEN, STK11, NF1, and CDH1 when individual features or the family history indicated. Since that time testing has increasingly been carried out using next-generation sequencing panels of a much larger series of genes. Typically, commercial companies offer a breast cancer panel and wider panels that include genes that have often never been linked convincingly (see above spurious section). In 2015 Easton et al. [99] suggested limiting breast cancer panels to genes that at least demonstrated a 2-fold OR of breast cancer. At the time only BRCA1, BRCA2, PALB2, ATM, CHEK2, and TP53 met the strict case-control qualifications, with syndromic genes only qualifying based on cohort studies. The article used the Ambry Genetics online tool (https://www.ambrygen.com/providers/resources/prevalence-tool; Table 2). The top 5 genes are the most frequent in all of the panel studies reported [40]. The higher apparent rates from the Lynch syndrome mismatch repair genes PMS2, MSH6, MLH1, and MSH2 have been reported in a number of studies, but none show evidence of an increase in the unbiased Prospective Lynch Syndrome Database (PLSD) [100]. This may reflect bias in ascertainment towards families with additional cancers in the individual or family consistent with Lynch syndrome such as colorectal cancer. Controls from the BRIDGES study may also not be matched to the population and excluded missense variants and CNV which inflate the OR for many of the genes but especially PMS2 and MSH2 [101]. Although the BRIDGES study did find borderline significance for MSH6, none of the other mismatch repair genes were confirmed [47]. We would suggest limiting testing to a panel of BRCA1, BRCA2, PALB2, ATM, CHEK2, PTEN, and TP53, leaving TP53 out if the patient is aged over 45 years and does not fulfill the Chompret criteria. CDH1 should only be tested if the case is lobular and there is a family or personal history of gastric cancer or lobular breast cancer [43]. This will limit the number of variants of uncertain significance while identifying the vast majority of actionable genes. There could be a case for adding BARD1, RAD51C, and RAD51D for triple-negative breast cancer [47].
Interaction of Genetic and Environmental Risk Factors
Past history may not be an adequate guide for future events. In a number of families for which data are available, the age of onset of cancer seems to decline over several generations and its frequency increases. This apparent trend might be explained by selection bias, but data from Iceland again suggest that this is real and can be attributed to the environmental and lifestyle factors that account for the rising incidence of breast cancer in populations in most economically advanced countries of northern Europe and North America. At present, most evidence suggests that standard risk factors such as reproductive history, breast-feeding, use of oral contraceptives or hormone replacement therapy, diet, alcohol consumption, or any other lifestyle factor also influences the cancer risk (penetrance) of carriers of breast cancer gene mutations. For example, the penetrance of the Icelandic BRCA2 founder mutation increased from 20 to 80% during the 20th century [102].
Contralateral Breast Cancer Risk (SNP and Known High-Risk Single Genes)
With improved breast cancer survival and high breast cancer incidence, some women are at an increased risk of developing a contralateral breast cancer. Whilst the overall risk is 0.4–0.5% per annum, estimates of likelihood are influenced by various factors including germline genetics [103].
Where a high-risk single gene disorder is present, prospective cohort studies have shown a contralateral breast cancer risk of ∼2% per annum for BRCA1 and 1–2% per annum for BRCA2 [11]. The lower incidence for BRCA2 likely reflects that the breast cancers occurring in BRCA2 mutation carriers tend to be estrogen receptor positive where therapeutic endocrine therapy is indicated. Oophorectomy, particularly undertaken at a younger age (< 45 years), is associated with a significant contralateral breast cancer risk reduction in BRCA2 carriers [10]. For the rarer, but high-risk, equivalent single gene alterations (Fig. 2), prospective contralateral breast cancer risk data is lacking, although a pragmatic BRCA1/2 equivalent high-risk approach is offered in the clinic. Nonetheless, for TP53 carriers diagnosed at age < 35 years, contralateral risks appear higher than for BRCA1 and BRCA2 [104] and there is evidence of a relatively high rate of contralateral breast cancer for the moderate-risk genes ATM and CHEK2 and a higher frequency of synchronous bilateral disease [105, 106].
Considering that collectively SNP account for a greater proportion of the familial risk of breast cancer, on a breast cancer population basis, it is those with women with a higher-risk PRS profile who are more likely to contribute to the population of contralateral breast cancers. For an SNP profile based on 313 variants, the contralateral lifetime breast cancer risk ranges from 12 to 20%, depending on the initial risk percentile [107].
Germline Genetics and Novel Therapeutic Strategies
Alongside the expansion of genetic testing capabilities has been the development of, and subsequent clinical trials involving, poly (ADP-ribose) polymerase (PARP) inhibitors, agents which render a cell unable to repair single-stranded DNA breaks. Where there is also defective homologous recombination to repair double-stranded breaks, cell lethality results. These agents are prime candidates for treatment of advanced breast cancers associated with germline or acquired mutations of BRCA1/2 and potentially also PALB2 given their functional roles in homologous recombination pathways. With advances in genomic technologies enabling improved turnaround times and testing being offered in mainstream clinical settings, identification of a germline BRCA1/2 mutation in the oncology setting has important therapeutic implications, with PARP inhibition of advanced breast cancers being associated with increased progression-free survival over standard care [108, 109]. Given their use now in a maintenance setting in ovarian cancer, they well be employed after primary treatment even for earlier-stage breast cancer.
Conclusion
Breast cancer predisposition is complex, being influenced by multiple genetic and environmental factors, and our knowledge of the genetic predisposition landscape has changed markedly over the past 30 years. Thus, PV of BRCA1 and BRCA2 are more common than originally thought, and the associated cancer risks have been shown to be influenced by both environmental factors and lower-risk susceptibility alleles (SNP). While considerable effort has been made toward the identification of further breast cancer predisposition genes, collectively known SNP and currently unidentified genetic modifiers of risk are likely to account for the remaining heritability. With advances in genomic technologies, more widespread genetic testing is now available but needs to be concomitant with accurate variant interpretation and family follow-up. Looking to the future, the challenges that lie ahead will include the incorporation of low-risk allele detection in the clinic and accurate risk stratification so that cancer prevention and early detection strategies can be put in place, especially for contralateral risk in newly identified cases.
Conflict of Interest Statement
D.G.E. has undertaken consultancy work for Astrazeneca and Springworks.
Funding Sources
D.G.E. and E.R.W. are supported by the National Institute for Health Research (NIHR) BRC Manchester (grant reference No. 1215-200074).
Author Contributions
All of the authors contributed to all aspects of this article.

