

Abstract
Background: Longitudinal bone growth is regulated by multiple endocrine signals (e.g., growth hormone, insulin-like growth factor I, estrogen, and androgen) and local factors (e.g., fibroblast growth factors and their receptors and the C-natriuretic peptide/natriuretic peptide receptor-B pathway). Summary: Abnormalities in both endocrine and local regulation of growth plate physiology cause many disorders of human skeletal growth. Knowledge of these pathways creates therapeutic potential for sustaining or even augmenting linear growth. Key Message: During the past 4 decades, advances in understanding growth plate physiology have been accompanied by development and implementation of growth-promoting treatments that have progressed in both efficacy and specificity of action. This paper reviews the history and continuing evolution of growth plate therapeutics.
Introduction
Growth Plate Physiology
The growth plate is the cartilaginous portion at the ends of long bones where longitudinal growth takes place. This region is characterized by high metabolic activity under control of a variety of hormones and signaling compounds [1]. Longitudinal bone growth is a complex multifactorial process of endochondral ossification in which cartilage is first formed and then remodeled into bone tissue. The postnatal growth plate contains chondrocytes at different stages of differentiation and separated into 5 layers (Fig. 1). In a coordinated stepwise process, the chondrocytes pass through a resting -, proliferative -, pre-hypertrophic -, hypertrophic -, and terminal phase. The resting zone chondrocytes, farthest from the primary ossification center, replicate at a slow rate and act as stem-like cells that replenish the pool of proliferative chondrocytes. Resting zone chondrocytes produce a “growth plate-orienting factor” that instructs the proper spatial orientation of adjacent proliferative chondrocytes. As cells within the resting zone divide, the proliferative zone is formed in which chondrocytes replicate at a high rate, become arranged into columns, and contribute to bone elongation. Hypertrophic chondrocytes generated from terminal differentiation of proliferating zone chondrocytes enlarge in columns parallel to the axis of elongation. Cell swelling during chondrocyte hypertrophy enables chondrocytes within the growth plate to enlarge rapidly. This phase of endochondral ossification, during which chondrocytes increase their height about 6- to 10-fold, serves as the major factor regulating the growth rate among endochondral bones. Hypertrophic chondrocytes calcify surrounding extracellular matrix and produce factors that attract bone cell precursors, bone cells, and blood vessel growth, and undergo apoptosis shortly before the blood vessels invade the chondrocyte lacuna. The overall effect of this process of chondrocyte proliferation, hypertrophy, and extracellular matrix secretion is elongation of bones and progressive creation of new bone tissue at the bottom of the growth plate.
![Fig. 1. Hormonal regulation of the growth plate (modified from [1, 17]).](https://karger.silverchair-cdn.com/karger/content_public/journal/hrp/94/9-10/10.1159_000520812/1/m_000520812_f01.jpeg?Expires=1716288440&Signature=nqAw9tBCeE8QFhUxfbewU~afOcuuR8CnCJZFToHnlmp6JrOdaSVmsTLSPrQ2WaOocG4lYMKUCSlalDgr74KtsTIl17nG363hxdca0w-eM2QMEWEFz4ZaAnrLRQHN-QbnH-vSe6KYcG4C95VZAw1mn2wElqWGIo2Euxu3TlE-Ym5eq1IklgFYsNp1ps0JVtFmWENOv~ZgXGSOWwRpw2s~uM8byxQl5802FOlVoSCdNNWJsmCW-~SuKKvEjf7169S8KjzeZcbMvzDCLeQR6Ng~6i01hwgCAJY8JuTUDKQpQ6gPwyC2~gO8UpKyKfl7xm985pJ2DCWU-dU6Y10z2jnlEQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
With age, the rate of longitudinal bone growth declines, caused primarily by a decrease in chondrocyte proliferation associated with other hormone-independent structural, functional, and molecular changes termed growth plate senescence. Evidence suggests that growth plate senescence occurs because the progenitor chondrocytes in the resting zone have a limited replicative capacity – gradually exhausted with increasing cell division [2]. When the proliferative capacity of stem-like cells in the resting zone is exhausted, and in the presence of sex hormones, growth plate cartilage becomes completely replaced by bone. This event is termed epiphyseal fusion. This review focuses on hormonal regulation relevant to current and developing therapeutic interventions.
Hormone Regulation of Growth
While chondrocytes control the local processes of bone growth, they are in turn controlled by numerous regulatory factors that control the rate of proliferation, maturation, and ultimately the length and strength of the bone (Fig. 1). Growth hormone (GH) initiates and maintains the process of growth through the stimulation of insulin-like growth factor-I (IGF-I). According to the dual-effector hypothesis, GH promotes recruitment of resting chondrocytes into a proliferative state and stimulates local production of IGF-I, which then acts in a paracrine/autocrine fashion to increase chondrogenesis. The IGF-I signaling pathway plays an important role in promoting complete hypertrophic chondrocyte formation [3]. These effects are predominantly due to growth plate-generated IGF-I, and IGF-I deficiency results in pre- and post-natal growth retardation marked in the growth plate by disorganized columnar chondrocytes, decreased cell proliferation and cell hypertrophy, increased apoptosis, and delayed vascular invasion.
Estrogen influences longitudinal growth primarily and indirectly by augmenting GH secretion during puberty, but also through both growth-enhancing and -attenuating direct actions on the growth plate [4]. A direct effect of estrogen is to advance growth plate senescence causing proliferative exhaustion, and thus epiphyseal fusion. An important mediator of this growth plate closure process is vascular endothelial growth factor, the production of which is stimulated by estrogen in both males and females. Androgens stimulate linear growth partly due to aromatization to estrogens within growth plate cartilage [5], but also by direct interaction with androgen receptors on growth plate chondrocytes [6] – explaining the GH- and IGF-I-independent growth stimulating effects of nonaromatizable compounds such as dihydrotestosterone and oxandrolone [7].
Linear growth is slowed by deficiency of and accelerated by excess thyroid hormone. Hypothyroidism indirectly impedes linear growth by diminishing GH secretion and IGF-I, but also by decreasing chondrocyte proliferation and hypertrophy, slowing of vascular/bone cell invasion, and disruption of column organization [8]. Excess glucocorticoid (GC) slows longitudinal bone growth by inhibiting chondrocyte proliferation, hypertrophy, and cartilage matrix synthesis. Diminished GH secretion and/or altered IGF-I bioavailability have been described in some GC-treated patients [9, 10]. Slowing of growth plate senescence due to GC appears to explain the phenomenon of catch-up growth following transient GC exposure [11] and hypothyroidism [12].
Members of the FGF family of receptors (FGFRs) and their ligands are required for proper chondrocyte function, endochondral ossification, and overall skeletal development. Proliferative chondrocytes express FGFR3 and pre-hypertrophic/hypertrophic chondrocytes express FGFR1. These pathways inhibit the proliferation of chondrocytes, thereby limiting the longitudinal growth of bones. Thus, activating mutations in FGFRs impede linear growth and cause skeletal phenotypes such as achondroplasia and hypochondroplasia.
Accelerated linear growth and epiphyseal growth plate maturation in obese children, even in the setting of decreased GH production may be due to effects of increased insulin concentrations and activation of the insulin receptor in the growth plate. Leptin, increased in obese children, has direct effects on skeletal growth centers [13], enhancing chondrocyte proliferation and subsequent cell differentiation [14]. Leptin also increases growth plate aromatase activity [15], which along with estrogen produced through adipose tissue aromatization [16] accelerates skeletal maturation.
Parathyroid hormone (PTH)-related protein (PTHrP) supports chondrocytes and maintains the width of the growth plate. Mutations affecting PTHrP action (e.g., Gs-alpha) can result in a shortening of the proliferating zone, accelerated differentiation of hypertrophic chondrocytes, premature closure of the growth plate, and short stature [17]. Vitamin D facilitates normal linear growth indirectly by increasing intestinal calcium and phosphate absorption, but vitamin D metabolites produced locally in the growth plate also decrease the proliferation of chondrocytes through the PTHrP pathway. Thus, the full effect of vitamin D on the growth plate physiology is incompletely understood [1].
Growth Plate Therapeutics
Recombinant Human Growth Hormone
For many years, treatment of growth failure was confined to correction of underlying disease or hormonal disturbance. Following the advent of recombinant human growth hormone (hGH), however, treatment of growth disorders expanded beyond hormone-replacement to hormone augmentation therapy. Availability of hGH allowed investigation of the growth-promoting effects of pharmacological hGH in poorly growing children without GH deficiency (GHD) and led to multiple FDA-approved indications for its use [18].
Many children have intrinsic traits that predispose them to short stature despite normal hormonal function. These traits affect either the tempo at which growth is accomplished (e.g., in constitutional growth delay of growth and maturation) or the absolute growth potential of growth centers/plates (e.g., familial stature, intrauterine growth restriction, defined genetic syndromes or skeletal dysplasia).
Girls with Turner syndrome (TS) experience short stature with an average adult height without treatment of 143 cm (56 inches and ∼20 cm below mid-parental height [MPH]). Short stature is due primarily to haploinsufficiency for the short stature homeobox (SHOX) gene. Girls with TS were among the first to show that hGH supplementation could improve height velocity (HV) and height outcomes in non-GHD children [11]. Long-term follow-up in a controlled study showed that girls with TS treated with hGH achieved a mean increase of 7.1 cm in height and were on average 5.1 cm closer to MPH compared to the control group [19]. Additional observational studies have yielded better results with higher dosing of hGH [20]. Initiation of hGH treatment is recommended as early as 4 years of age at a dose of 45–50 μg/kg/day, increasing to as much as 68 μg/kg/day if adult height potential is substantially compromised [21]. Of note, such high doses can produce supraphysiological IGF-1 levels that have been epidemiologically associated with the incidence risk for breast [22] and colon malignancies in adults [23].
Children born small for gestational age (SGA) have a birth weight and/or length less than 2 SDS below the mean for their gestational age and sex (below the 3rd percentile). For the approximately 10% of these children who show persistent growth retardation at 2–4 years of age, treatment with hGH increases adult height attainment by a mean 1.3 SDS [24, 25]. Additional height gained with either intermittent or high-dose (up to 68 μg/kg/day; 0.48 mg/kg/week) hGH treatment is modest (∼0.5 SD or 1.25 in) compared to sustained lower dose treatment. Children with Russell Silver syndrome are often treated with hGH because of SGA (particularly when accompanied by hypoglycemia) and show a similar response to nonsyndromic children born SGA (mean total height gain ∼1.5 SDS) [25].
Idiopathic short stature (ISS) is a diagnosis of exclusion applied to short children with no identified etiology for poor growth. Recombinant hGH is also FDA-approved for treatment of children with ISS with height less than or equal to −2.25 SD below the mean (∼1st percentile), predicted adult height (PAH) below the normal range (160 cm [5 ft 3 in] for males, and 150 [4 ft 11 in] for females), and open epiphyses. Treatment of children with ISS with hGH at 55 μg/kg/day (0.375 mg/kg/week) can increase adult height, on average, by approximately 1 cm/year of hGH treatment [26]. Greater bone age (BA) delay and taller MPH predict greater response; otherwise, there are presently no additional clinical or biochemical determinants that reliably predict long-term response to hGH therapy prior to initiation of treatment [27].
Noonan syndrome (NS), an autosomal dominant disorder due most commonly to PTPN11 mutation, has short stature as a characteristic. Treatment of children with NS with hGH, FDA-approved at doses up to 68 μg/kg/day (0.48 mg/kg/week), increases near-adult height by a median 1.5 SD in males and 1.0 in females [28].
SHOX gene mutations occur in ∼1 in 1,000 newborns, and haploinsufficiency leads to reduced limb growth, short stature, and skeletal deformities [29]. Mutations in SHOX cause Leri-Weill dyschondrosteosis but are identified in approximately 2–8% patients with ISS [30, 31]. Treatment with hGH increases HV and adult height in SHOX haploinsufficiency patients [32]. The magnitude of this effect is similar to that observed in TS patients [33] and may be enhanced when combined with GnRHa initiated at the very start of puberty [34]. Disproportionate growth, with less robust growth of the lower extremities compared to trunk and arms, can occur [34].
Prader-Willi syndrome is characterized by short stature in addition to obesity, hypotonia, hypogonadism, cognitive impairment, and hypothalamic dysfunction which may cause variable GH secretion. Growth failure related to Prader-Willi syndrome is also an FDA-approved indication for hGH treatment in the USA, even in the absence of demonstrable GHD. Recombinant hGH treatment at 1.0–1.5 mg/m2/day (generally 30–35 μg/kg/day; 0.24 mg/kg/week) increases HV to a degree similar to children with severe GHD during their first year of hGH replacement therapy, and over 4 years, results in a cumulative mean change in height SDS of +1.8 ± 0.6 SDS [35].
Recombinant Human Insulin-Like Growth Factor-1
The first indication that IGF-I was of great importance in either endocrine or autocrine/paracrine actions at the level of the growth plate came from incubation experiments with costal cartilage obtained from hypophysectomized rats [36]. Daughaday et al. [37, 38] confirmed that a “serum sulfation factor,” now known as somatomedin-C or IGF-I, was the most important mediator of GHs effects on somatic growth. The original somatomedin hypothesis postulated that statural growth was regulated by GH via inducing “endocrine” IGF-I secretion by the liver, but evolved quickly when GH-stimulated local tissue IGF-I production for autocrine/paracrine action was found to be more contributory to somatic growth than hepatic IGF-I [39]. In addition, GH itself acts directly on the growth plate by promoting differentiation of cartilage precursor cells, while IGF-I promotes clonal expansion of these differentiated cells (the “dual effector theory,” as noted previously) [40], although in vivo studies seem to indicate that the actual mechanism is more complex than this theory would suggest [41, 42].
Recombinant human insulin-like growth factor-1 (rhIGF-1) was first administered to humans in 1989 and the efficacy and safety of rhIGF-1 therapy in patients with primary IGF-I deficiency (IGFD) has been evaluated for more than 3 decades [43]. Most of the therapeutic experience comes from observations in patients with severe short stature and a phenotype characteristic of GH receptor deficiency (Laron syndrome). Although most patients do not experience sufficient catch-up growth resulting in a height into the normal range, many patients treated with rhIGF-1 improve linear growth and adult height outcome compared with the growth expectations in the absence of IGF-I replacement [44]. rhIGF-1 is approved for severe forms of IGFD, defined as a height SD score ≤−3, and IGF-I SD score ≤−3 (in the USA), or IGF-I <2.5th percentile (in the European Union), all in the setting of normal or elevated GH concentrations. Even with the growth-promoting effects of rhIGF-1 treatment well-documented, the overall growth response in severe IGFD patients to long-term rhIGF-1 therapy remains below the growth response in GH-deficient patients treated with rhGH. This is in part because locally produced IGF-I is sub-optimally restored by the repletion of the endocrine IGF-I component and related to its short therapeutic half-life as concentrations of insulin-like growth factor binding protein-3 and the acid-labile subunit are low and unaltered by this therapy. Furthermore, the direct actions of GH on the growth plate in these GH insensitive patients remain absent during monotherapy with rhIGF-1. It is conceivable that newer formulations of IGF-1 will be developed in the future that will allow better delivery to the growth plate itself. For example, experiments with cartilage-targeting fusion proteins of IGF-1 that can deliver therapeutic molecules to the growth plate tissue have been shown to retain cartilage binding and IGF-I biological activity in both cell cultures and an animal model, thereby demonstrating potential for the therapeutic application in human growth disorders [45]. Since rhIGF-1 was approved, several neoplasms in IGF-1 treated pediatric patients have been reported, including a variety of malignancies. It is not known if there is a causative connection between therapy with rhIGF-1 and the development of such neoplasms. Although predominantly reported in short children with a rare genetic disorder – by itself known to increase the risk of malignancy, and reportedly after use of higher than recommended IGF-1 dosing, the findings precipitated a drug safety-related labeling change for rhIGF-1 in 2019 (www.accessdata.fda.gov/scripts/cder/daf).
Gonadotropin-Releasing Hormone Analogs
Gonadotropin-releasing hormone analogs (GnRHa) have been in use during the last 2 decades to improve height outcomes in children with different clinical presentations of central precocious puberty (CPP) [46]. These GnRHa products are altered forms of endogenous GnRH with an increased potency and longer half-life. The primary objective for CPP suppression with GnRHa is to avoid premature fusion of the growth plates, which could lead to reduced adult height potential. The first long-term trial with GnRHa therapy for CPP clearly demonstrated a decrease in HV and skeletal maturation and resulted in an improvement of the PAH [47]. The children treated in early trials presented with rapidly progressive CPP and were started on GnRHa early (mean age 5.2 years) [48]. Many other studies have confirmed the benefit of GnRHa therapy for (mainly idiopathic) CPP, if such treatment intervention occurs no later than 7–8 years of age [49]. It should be noted, however, that height gains achieved with GnRHa suppression are highly variable, as the difference between adult height outcome and PAH can range from +2 to +10 cm [50, 51]. The use of GnRHa to preserve adult height in (predominantly) girls with milder CPP is more likely to be of dubious benefit, because many of these girls, if left untreated, have a slow tempo of puberty, and their height in adulthood will, on average, not end up more than 4.4 cm (<2′′) below their target height [52]. Individuals with a history of being born SGA, or adopted from another country, or who have severe developmental delay, on the other hand, likely still benefit from GnRHa-mediated pubertal suppression, even at more advanced ages. Overall, GnRHa therapy is considered to be very safe [49]. Although it seems that bone mineral accrual during GnRHa treatment is suboptimal, bone density evaluated after stopping therapy and in late adolescence is in the normal range [53].
Androgens
By interacting with androgen receptors on growth plate chondrocytes [4], androgens exert GH- and IGF-I-independent growth stimulating effects [5]. Thus, growth-promoting therapies for short peri-pubertal boys include low-dose androgen therapy with either injectable testosterone (e.g., 50–100 mg monthly intramuscularly) or low-dose oral oxandrolone (e.g., 1.25–2.5 mg/day). In controlled trials, both increase HV rate by 3–5 cm/year [54, 55]. Testosterone esters are used primarily in boys with constitutional delay of growth and maturation to both increase HV and induce secondary sex characteristics. Adult height outcomes are reported as appropriate for family [56] and similar to untreated subjects [57], but adequately powered, long-term randomized controlled trials to confirm these observations are lacking.
To avoid accelerated estrogen-mediated epiphyseal maturation, oxandrolone (not aromatized to estrogen) is theoretically preferred over testosterone, particularly when skeletal age is <11 years. In contrast to testosterone, oxandrolone increases HV independent of changes in GH/IGF-1 production [58]. Oxandrolone is usually discontinued after a documented rise in endogenous testosterone [55]. Addition of oxandrolone to hGH in treatment of girls with TS also appears to increase their adult height attainment by a mean of approximately 4 cm [59]. Oxandrolone at very low dosages (e.g., 0.625–1.25 mg/day) has low risks (e.g., adverse hepatic or lipid effects), and the substantial cost-savings of oxandrolone versus hGH treatment for non-GHD short children balance favorably with small differences in height attainment [60]. Of note, however, oxandrolone is not available in certain countries.
Estrogen
Because of its bimodal influence on the growth plate (i.e., enhancing GH secretion at low levels and advancing growth plate senescence and epiphyseal closure at higher levels), estrogen has been investigated for both growth-enhancing and growth attenuating purposes. Low-dose estrogen treatment accelerates growth in both prepubertal boys and girls [61], but investigation of possible adult height increase is confined to girls with TS, in whom supplementation of hGH treatment with ultra-low-dose ethinyl estradiol (25–50 ng/kg/day) during childhood modestly increased adult height (about 2 cm) [62]. In the past, high-dose estrogen treatment to reduce final height of healthy tall girls was widely used. In this group, treatment with oral administration of estradiol (2.5–20 mg/day) or ethinyl estradiol (0.05–0.3 mg/day) resulted in an estimated ratio of bone-age to height-age advancement between 2.0 and 3.7 [63]. Reported reductions in adult height, however, were typically only 2–7 cm and inversely proportional to bone age at the start of estrogen exposure, with a predicted growth-attenuating effect of 6 ± 1.5 cm at a bone age of 10 years, 3 ± 1.0 cm at a bone age of 12 years, and no effect at a bone age of 14 years [64]. Importantly, follow-up studies reported a dose-related increased risk of reduced fecundity and infertility in women who received such treatment [65, 66]. Increased social acceptance of tall stature in females and the greater visibility of positive tall female role models have made requests for this treatment a rare occurrence presently. Criteria for initiation of growth-attenuation therapy have changed accordingly [67, 68], and estrogen is currently most often used to limit growth in children with severe physical and cognitive impairment for whom the prospect for excess height is viewed as a disability [69-72].
Aromatase Inhibitors
Aromatase is a cytochrome P450 enzyme that catalyzes conversion of androgens to estrogens. Aromatase gene variants are evidence that estrogen has an important role in bone maturation and closure of growth plate [73]. Aromatase inhibitors (AI) have been used for treatment of estrogen-sensitive breast cancer, gynecomastia, and peripheral precocious puberty, specifically in McCune-Albright Syndrome [74]. Anastrozole and letrozole are third-generation nonsteroidal AIs that can bind reversibly with high potency to the cytochrome P450 to decrease conversion of androgens to estrogen, which can decelerate growth plate fusion by minimizing estrogen action. Because they are given orally at relatively low cost, AIs have become a treatment option for boys with short stature to increase adult height [75]. In girls and premenopausal women, AIs are contraindicated unless used for specific diagnoses (e.g., McCune Albright syndrome) since inhibition of estradiol synthesis leads to a net decrease in bone deposited at a crucial age of bone accrual [76] and reduced estradiol-mediated negative feedback of the hypothalamic-pituitary-gonadal axis leads to gonadotropin-stimulated increased ovarian androgen production.
In 52 adolescent boys with GHD treated with rhGH randomized to co-treatment with anastrozole or placebo, bone age advancement was significantly slower in the anastrozole group compared to placebo, resulting in a net increase of PAH in the combination group of 6.7 cm ± 1.4 cm compared to the placebo plus GH treated group (only 1.0 cm in 36 months) [77]. Free fat mass accrual was greater with combined therapy, and measures of bone health showed no detrimental effects following AIs treatment [78]. A double-blinded, placebo-controlled study of 31 boys with ISS treated with letrozole 2.5 mg/day showed an increase in PAH of 5.9 cm (p < 0.0001) and a BA SDS increase of +0.7 (p < 0.0001) compared to the placebo group [79]. An additional double-blind, placebo-controlled study performed in a group of 91 boys with constitutional delay of puberty and predicted short stature showed a significant increase in PAH after 2 years of letrozole compared to patients treated with oxandrolone or placebo [80].
Not all studies, however, show a positive effect of AI treatment in increasing height in individuals with ISS. A retrospective chart review of 20 boys with a bone age of ≥13 years and short stature treated with anastrozole for 18–30 months, showed only a small, nonsignificant increase in PAH [81] and a similar retrospective review reported no significant change in PAH with either anastrozole or letrozole treatment [82]. In addition to limited strong evidence for long-term efficacy, questions still remain regarding long-term safety. AI treatment both markedly decreases estradiol levels and increases testosterone, follicle-stimulating hormone, and luteinizing hormone levels. Effects of high testosterone levels, sometimes above the adult male reference range, have not been studied long-term. In boys, limited available data indicate that as puberty progresses, bone density increases similarly in patients receiving AIs and in control subjects [83].
Thus, while AIs have been used to treat short stature in males for more than 20 years, caution should still be taken before prescribing this intervention. Additional data regarding bone health, reproductive function [84], lipid [80] and carbohydrate metabolism, and adrenal function are needed to fully assess the benefits and burdens of AI therapy [85].
Emerging and Future Growth Plate Therapeutics
Long-Acting Growth Hormone
Treatment with limited supplies of pituitary derived GH typically entailed intramuscular injection given 3 times weekly, whereas recombinant hGH therapy is administered as a daily subcutaneous injection since its approval in 1985. Numerous studies have shown that adherence and persistence influence growth outcomes. The burden of daily injections can lead to treatment fatigue and nonadherence, particularly in adolescents [86]. Measurement of adherence using patient report, electronic device capture, pharmacy refill data, and returned vial count have shown that nonadherence ranges from 21% to 70% [86-91] and is associated with a reduction in HV. Because reduction in frequency of medication administration may improve adherence, development of long-acting growth hormone (LAGH) preparations has been an area of keen investigation.
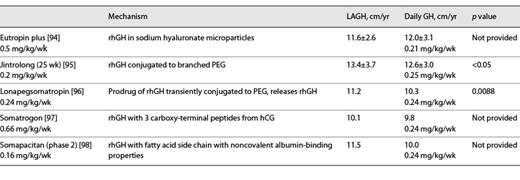
Interest in long-acting forms of GH predated the development of recombinant hGH (Fig. 2) [92], as gel encapsulation was studied as a method of prolonging the release of pituitary GH [93]. The first form of LAGH (Nutropin DepotTM or hGH encapsulated in biocompatible, biodegradable, polylactide-coglycolide polymer microspheres) was approved in 1999 for once or twice monthly intramuscular injections but was discontinued in 2004 due to manufacturing issues. Since then, multiple methodologies have been used to prolong the biological availability of hGH [92]. Some early preparations focused on prolonging the presence of GH in the subcutaneous tissue caused local lipoatrophy. Currently, 2 subcutaneous depot preparations are approved for clinical use in children with GHD. Eutropin Plus® (a sustained release formulation of hGH contained in sodium hyaluronate microparticles) is approved in South Korea as a once weekly subcutaneous injection (Table 1) [94]. Jintrolong® (a peg-ylated 62 kDa GH) was approved in 2014 for treatment of pediatric GHD in China as a once weekly subcutaneous injection (Table 1; Fig. 2) [95].
![Fig. 2. Timeline of GH treatment development (adapted from [92]). EU, European Union; Pit-hGH, pituitary-derived human growth hormone; PGHD, pediatric growth hormone deficiency.](https://karger.silverchair-cdn.com/karger/content_public/journal/hrp/94/9-10/10.1159_000520812/1/m_000520812_f02.jpeg?Expires=1716288440&Signature=JcBOdjKMy9ypKugzvVyteGwhk99KveQwyOx1lG6QhM7LaA8Y3WtFq~U2gJhi80L8Q7tmEopRK-YlO6o9IcTPYpwb2jU6EpD2NgdPwfDjnkW--I850y~1T768H7BeiDAhKHZWh1h2MgurTUjb2IFlHH5q3ihe6OC-1KPul-4o6lLpbg514JVZYT72tilxViRXPOUgF9MIcVMFCorJzMgq-Q9kTllK4UMkGmMpPZu1XqU6E1f5ghDF4waer-yArYtqovtMaWFb0dS487YjhqPR0OD0HdC2dT2y4oJMFUdISdsI07wFvzipA4gqmPpQXoR7ZbMLAJF9Njv~dUUq0nHGqQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Timeline of GH treatment development (adapted from [92]). EU, European Union; Pit-hGH, pituitary-derived human growth hormone; PGHD, pediatric growth hormone deficiency.
Timeline of GH treatment development (adapted from [92]). EU, European Union; Pit-hGH, pituitary-derived human growth hormone; PGHD, pediatric growth hormone deficiency.
To date, there are 3 LAGH preparations in clinical trials for treatment of pediatric GHD in the USA and Europe. These LAGH preparations prolong half-life through either formation of a prodrug that releases native GH (lonapegsomatropin, Skytrofa®), increased binding to serum albumin (somapacitan, Sogroya®) or formation of a fusion protein with known sequences from other circulating proteins (somatrogon). Phase 3 clinical trials of once weekly lonapegsomatropin show superior HV and equal safety profile in children with GHD compared to those receiving daily somatropin at the same weekly dose (Table 1) resulting in FDA approval on August 25, 2021 [96]. Phase 3 clinical trials of once weekly subcutaneous administration somatrogon show noninferior HV in children with GHD compared to those receiving daily somatropin (Table 1), with similar safety profiles [97]. The clinical data have been submitted to the regulatory authorities and is pending response expected in late 2021. Phase 2 clinical trials of once weekly subcutaneous administration with somapacitan (0.16 mg/kg/week) show superior HV in children with GHD compared to those receiving daily somatropin (0.24 mg/kg/week) [98].
It will be necessary to determine the best methodology for LAGH dose adjustment [28]. Measurement of IGF-I and IGFBP-3 has been used for dose adjustment and safety monitoring of hGH treatment. Whereas serum IGF-I in children receiving daily hGH is stable throughout the day, a peak to trough pattern of IGF-I will follow a once weekly LAGH injection, requiring that the provider know the date and approximate time of the injection to properly interpret IGF-I values obtained.
While intuitively attractive, it remains to be proven that LAGH treatment will improve adherence compared to daily hGH. In addition, for some children, LAGH may not offer benefits compared to daily hGH; e.g., children with severe GHD could be at risk of hypoglycemia during the trough period following LAGH injection. And while risks of transient elevations of GH and IGF-I for tumor development with daily GH are low, more long-term data are needed regarding safety of LAGH treatment for cancer survivors.
GH Secretagogues
Normal GH secretion involves pulsatile release from the pituitary in response to growth hormone releasing hormone with the largest pulses occurring during sleep. In children with partial GHD, the frequency and amplitude of GH pulses are reduced. In such patients with an intact hypothalamic-pituitary axis, GH secretagogues promote secretion of growth hormone releasing hormone, GH, or both in a pattern potentially more physiological than injected hGH. An intact hypothalamic-pituitary axis may also allow enough feedback by IGF-I to avoid excessive GH secretion and elevated IGF-I. Two GH secretagogues are currently in development for diagnostic and/or therapeutic indications in children.
Macimorelin is an agonist at the GH secretagogue receptor 1a, also known as the ghrelin receptor. It is currently FDA-approved as an oral diagnostic agent for GHD in adults and the use for pediatric GHD is currently being investigated. LUM-201 (ibutamoren), also an agonist at the GH secretagogue 1a/ghrelin receptor, is under investigation as an oral therapeutic agent for children with GHD. In adults, LUM-201 consistently increased GH secretion and IGF-I over the course of 1 year. A predictive enrichment marker incorporating low pretreatment IGF-I and ability to increase GH secretion in response to LUM-201 may identify children likely to both respond and not respond to LUM-201. It is estimated by some that about 60% of children with GHD will respond to this GH secretagogue with improved GH secretion [99, 100] and increased HV [99]. A phase 2 clinical trial of LUM-201 is comparing growth rates during treatment with different doses of LUM-201 to daily hGH over 6 months in children with GHD. Future clinical trials will determine whether GH secretagogues are safe and effective therapeutic options in children with GHD and other growth disorders.
FGF and CNP Path Modulators
Rare disease drugs that target the growth plate to stimulate chondrocyte differentiation and hypertrophy, including analogues of C-type natriuretic peptide (CNP), FGFR3-selective tyrosine kinase inhibitors, anti-FGFR3 antibodies, and soluble forms of FGFR3, are possible treatments for achondroplasia and other causes of short stature (Fig. 3). CNP is a small peptide and potent stimulator of endochondral ossification. CNP binds to its receptor, natriuretic peptide receptor-B (gene NPR2) causing transformation of GTP (guanosine 5′-triphosphate) into cGMP (cyclic guanosine monophosphate). In the growth plate, this binding of CNP to NPR-B stimulates chondrocyte differentiation and hypertrophy through downstream signaling by inhibiting mitogen-activated protein kinase (MAPK) signaling. This leads to changes in gene expression in the chondrocytes and increases formation of extracellular matrix. Pathogenic variants that result in overexpression of CNP in humans are associated with enhanced skeletal growth leading to abnormally tall stature. Transgenic mice with systemic or cartilage-specific overexpression of CNP show skeletal overgrowth phenotype while CNP and NPR-B knockout mice exhibit growth failure due to impaired endochondral bone growth [101]. In several families with NPR2 heterozygous variants, mild disproportionate short stature and/or phenotypic or radiographic indicators are present, while homozygous mutations in NPR2 lead to skeletal dysplasia with significant short stature and body disproportion [102, 103].
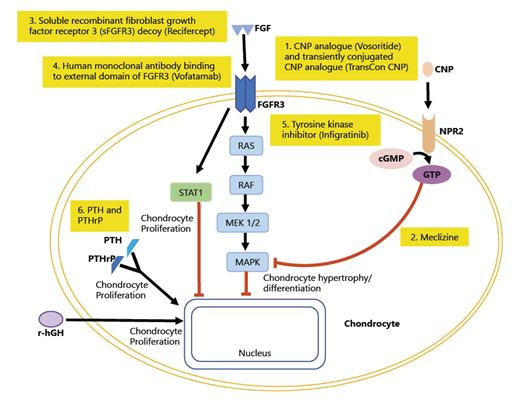
FGF and CNP path modulators. At the growth plate of chondrocyte, FGF acts on FGFR3 which activates RAS-MAPK pathway resulting in the inhibition of chondrocyte proliferation and differentiation. CNP binds to its receptor, gene NPR2 causing transformation of GTP into cGMP leading to inhibition of MAPK leading to chondrocyte hypertrophy and differentiation. rhGH leads to chondrocyte proliferation. There are drugs currently considered under investigation or potential therapeutic agents at the level of the growth plate: (1) CNP analogue (Vosorotide) and transiently conjugated CNP analogue (TransCon CNP). (2) Meclizine inhibits MAPK pathway leading to increased chondrocyte hypertrophy and differentiation. (3) sFGFR3 (Recifercept) is a soluble recombinant human FGF receptor that binds to FGF and reduces its effect on the growth plate. (4) Human monoclonal antibody binding to external domain of FGFR3 (Vofatamab) is a drug that inhibits receptor FGFR3. (5) Tyrosine kinase inhibitor (Infigratinib) has selectivity for the FGFR receptor family and, by inhibiting FGFR3 phosphorylation, decreases expression to a normal level of activity in chondrocytes. (6) PTH and PTHrP lead to chondrocyte proliferation.
FGF and CNP path modulators. At the growth plate of chondrocyte, FGF acts on FGFR3 which activates RAS-MAPK pathway resulting in the inhibition of chondrocyte proliferation and differentiation. CNP binds to its receptor, gene NPR2 causing transformation of GTP into cGMP leading to inhibition of MAPK leading to chondrocyte hypertrophy and differentiation. rhGH leads to chondrocyte proliferation. There are drugs currently considered under investigation or potential therapeutic agents at the level of the growth plate: (1) CNP analogue (Vosorotide) and transiently conjugated CNP analogue (TransCon CNP). (2) Meclizine inhibits MAPK pathway leading to increased chondrocyte hypertrophy and differentiation. (3) sFGFR3 (Recifercept) is a soluble recombinant human FGF receptor that binds to FGF and reduces its effect on the growth plate. (4) Human monoclonal antibody binding to external domain of FGFR3 (Vofatamab) is a drug that inhibits receptor FGFR3. (5) Tyrosine kinase inhibitor (Infigratinib) has selectivity for the FGFR receptor family and, by inhibiting FGFR3 phosphorylation, decreases expression to a normal level of activity in chondrocytes. (6) PTH and PTHrP lead to chondrocyte proliferation.
Fibroblast growth factor (FGF) acts on FGF receptor 3 (FGFR3) causing activation of RAS and MAPK pathways (Raf-MEK-ERK), which causes inhibition of chondrocyte proliferation and differentiation. This leads to decreased gene expression of certain genes in chondrocyte nucleus that leads to decreased formation of extracellular matrix. Achondroplasia is the most common form of disproportionate short stature due to an autosomal dominant mutation in the FGFR3 gene that constitutively activates the MAP kinase extracellular signal-regulated kinase pathway in chondrocytes, which inhibits endochondral ossification.
Once-daily subcutaneous administration of recombinant CNP analogue, vosoritide, promoted long-bone growth in juvenile, skeletally normal mice and monkeys and corrected the dwarfism phenotype in mice with achondroplasia [104]. This led to clinical trials in patients with achondroplasia. In a multinational, phase 2 trial, a total of 35 children between age 5 years and 14 years of age with achondroplasia were enrolled to receive vosoritide once-daily subcutaneously at different dosages. The study demonstrated the side-effect profile generally mild and there was a sustained increase in the annualized HV for up to 42 months [105]. This led to the phase 3, randomized, double-blind, placebo-controlled trial in children age range 5 to <18 years with achondroplasia with 60 assigned to receive 15.0 μg/kg/daily dose of vosoritide and 61 to receive placebo. The adjusted mean difference in annualized HV between patients in the vosoritide group and placebo group was 1.57 cm/year in favor of vosoritide compared to placebo (95% CI [1.22–1.93]; 2-sided p < 0.0001). The study showed that there were no serious adverse events related to treatment and no deaths occurred [106].
It is still unclear how this relatively modest increase in growth rate with vosoritide treatment will affect adult height attainment in children with achondroplasia. The study discussed above has been extended to an open-label study (NCT03424018) to follow these children until they reach adult height and to assess changes in medical complications associated with achondroplasia, functional outcomes, quality of life, and increase growth velocity during puberty. Additional clinical trials (NCT03583697, and NCT04554940) for ages 3–60 months aim to determine whether vosoritide treatment can decrease foramen magnum stenosis, which can lead to brainstem compression and sudden death [107]. Vosoritide was recently approved for treatment of achondroplasia by European Commission (EC) and is currently awaiting FDA approval. TransCon CNP, a transiently conjugated CNP analogue, is currently being studied in a placebo-controlled Phase 2 clinical trial (NCT04085523) in prepubertal children with achondroplasia aged 2–10 years receiving escalating doses as a once weekly subcutaneous injection.
Soluble recombinant human FGF receptor 3 (sFGFR3), or Recifercept, is another drug under clinical investigation for patients with achondroplasia. sFGFR3 binds to FGF isoforms in vitro and in cellular model systems and reduces FGFR3 signaling. It sequesters FGFR3 ligands subsequently normalizing activation of the mutated FGFR3 receptor, essentially acting as a decoy with the extracellular domain of FGFR3. In transgenic mouse model of achondroplasia, Recifercept restored reduced body weight and long bone growth [108]. At this time, there is a phase 2 randomized, 3-arm parallel group dose study (NCT04638153) of Recifercept on its way to study safety, tolerability and efficacy in children with achondroplasia.
There are other drugs that may potentially function at the growth plate. Meclizine, an antihistamine drug used for travel sickness blocks ERK1/2-MAPK pathway along with inhibiting FGFR3 activity. In mice studies, meclizine increased length of bones and strengthened trabecular bone. There has been only been a phase Ia trial completed on patients with achondroplasia, showing that it did not lead to any significant side effects. A human monoclonal antibody that binds to external domain of FGFR3, Vofatamab, that is under investigation for multiple myeloma has been considered also, but may be too large to penetrate the extracellular matrix. PTH and PTHrP act on the growth plate at the PTH/PTHrP type 1 (PPR) receptor to proliferate and differentiate chondrocytes; however, long-term effects of PTH are not known and safety of recombinant PTH needs to be studied. Infigratinib (NVP-BGJ398/BGJ398) is an orally administered tyrosine kinase inhibitor which has selectivity for the FGFR receptor family and by inhibiting FGFR3 phosphorylation, decreases expression to a normal level of activity in chondrocytes. In mouse models, treatment with the NVP-BGJ398 inhibitor caused elongation of the femur, alleviated the disruption of chondrocyte differentiation, increased limb growth, and corrected lumbar vertebra size [109]. Finally, fusion of single-chain human cartilage-targeting single-chain antibody fragments (CaAb) with growth stimulating factors (in addition to IGF-1 mentioned above) has the potential to provide on-target efficacy of therapeutic agents at the growth plate with less off-target systemic effects [45].
Conclusion
Axial elongation of long bones via endochondral ossification is a highly complex process tightly controlled by systemic hormones, local growth factors, signaling cytokines, and cellular differentiation. Accordingly, numerous genetic defects, diseases, deficiencies, and therapeutic agents can interfere with growth, and alternatively, several pathways offer theoretical opportunities for sustaining or even augmenting growth. During the past 4 decades, progress in understanding of normal growth plate physiology has been accompanied by development and implementation of growth-promoting treatments that have evolved in both efficacy and specificity of action. Further elucidation of mechanisms controlling normal and abnormal growth promises to lead to even more specifically targeted, safe, and effective growth plate therapeutics.
Acknowledgment
The authors thank Ron Rosenfeld, MD, for his careful review of and suggestions for the manuscript.
Conflict of Interest Statement
Dr. D.B. Allen has received honoraria from BioMarin Pharmaceutical. Dr. N. Merchant has no personal financial disclosure. She is co-investigator for Vosoritide Trial for Selected Genetic Causes of Short Stature. Dr. B.S. Miller is a consultant for Abbvie, Ascendis Pharma, BioMarin, EMD Serono, Novo Nordisk, Orchard Therapeutics, Pfizer, Sandoz, Sanofi Genzyme, Tolmar, and Vertice and has received research support from Alexion, Abbvie, Amgen, Ascendis Pharma, Lumos Pharma, Novo Nordisk, OPKO Health, Pfizer, Sandoz, and Tolmar. Dr. P.F. Backeljauw is currently a consultant for, or has received honoraria from, Novo Nordisk, Novartis/Sandoz, Ascendis Pharma, BioMarin, Endo Pharmaceuticals, Jupiter Bioventures and Ipsen, and receives research support from Novo Nordisk and Ipsen.
Funding Sources
No funding was received for this study.
Author Contributions
Each author has made substantial contributions to the conception or design of the work, or to the acquisition, analysis, or interpretation of data for the work; participated in drafting the work or revising it critically for important intellectual content; approved the final version to be published; and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
