

Abstract
The last decade has been a remarkably productive one in the field of bone biology. New insights into the maintenance of a normal bone microenvironment have led to significant advances in our understanding of many important skeletal disorders, including renal osteodystrophy. Novel targets for therapeutic manipulation have been exposed and encouraging progress made towards new treatments. In addition, just as clinical studies have alerted us to the potential hazards of vascular calcification, basic science has unearthed the intimate nature of the relationship between the previously separate disciplines of bone and vascular biology. The clinical nephrologist, however, may be forgiven a little cynicism at this point. If such progress has been made, why do the same proverbial difficulties confront us in day-to-day practice? Control of phosphate remains inadequate, despite a literature which constantly reaffirms its crucial importance, and parathyroid hyperplasia seems inevitable in many patients. Furthermore, even the satisfaction of successful control of serum parathyroid hormone concentration must now be tempered by disquiet regarding the skeletal and cardiovascular consequences of oversuppression. This review aims to provide an update of the latest developments in relevant skeletal research and to assess how recently acquired knowledge may improve clinical nephrological practice over the next five years.
Introduction
Characterisation and Diagnosis of Osteodystrophy
Bone is not static; it continuously adapts to prevailing mechanical and metabolic requirements by a dynamic remodelling (and in children modelling) process centred on the coupling of osteoblastic formation and osteoclastic resorption. Influence on remodelling is exerted by both ‘classical’ osteotropic hormones, i.e. parathyroid hormone (PTH), calcitriol, sex steroids and thyroid hormones, and many locally produced cytokines and growth factors. Disturbances of this process impair bone quality and quantity. Renal osteodystrophy is a complex and heterogeneous disorder best thought of in terms of a histomorphometric continuum, with high and low turnover disease at opposing ends. High turnover disease is closely associated with the clinical entity of secondary hyperparathyroidism (SHPT). It is characterised by increased numbers of both osteoblasts and osteoclasts, and in many cases by the formation of poor-quality ‘woven’ bone. With low turnover disease, reduced numbers of bone cells, very low rates of bone formation and serum PTH that is in, or just above, the normal range are characteristic. The adynamic bone disorder, adynamic bone disease (ABD) and aplastic bone disease are all terms that have been applied to this scenario. Confusion can arise because high and low turnover states are not mutually exclusive – they may co-exist in bone biopsy specimens. In addition, interchange from one form to another is common and often driven by current treatments. This can be difficult to conceptualise because we are inclined to think in terms of an absolute value of serum PTH (we deal with these every day), rather than within a histological framework (we rarely perform bone biopsies). A key challenge for the future is to refine our ability to both diagnose and subcategorise bone pathology without over-reliance on the serum PTH or recourse to bone biopsy.
In this regard, the utility of several biochemical markers of bone turnover to predict the type of skeletal lesion has come under scrutiny. Elevations in osteoblast-derived bone-specific alkaline phosphatase correlate well with PTH and are predictive of high turnover disease, though less useful for the differentiation of low from normal turnover [1]. Other osteoblast products, particularly osteocalcin, and various breakdown products of type I collagen, including pyridinoline and deoxypyridinoline, have also been studied and could, eventually, play a larger diagnostic role as assays and interpretation are refined in relation to histology. In the case of PTH itself, it is increasingly clear that in addition to ‘intact’ PTH, which acts on its targets via the classical PTH-1 receptor, most commonly utilised first-generation immunometric assays also detect other biologically active fragments of the hormone. These N-terminally truncated peptides appear to exert an inhibitory influence on bone cells, almost certainly via a distinct C-PTH receptor, and could be a contributory factor to perceived skeletal resistance to PTH [2, 3]. Second-generation immunometric assays, more specific for ‘whole’ PTH (1-84) molecule, have been developed and are likely to attract increasing attention as their clinicopathological validation progresses.
At present, imaging plays only a minor role, with plain X-rays increasingly redundant in the diagnosis of osteodystrophy, though of increasing importance in the assessment of vascular calcification. The application of DEXA technology for the assessment of bone mineral density in the uraemic patient is complicated. There is, as yet, no consensus on the optimum site(s) of measurement and correlations with both histology and fracture risk are poor. For these reasons, estimates of bone mineral density are best viewed as part of an overall data set, rather than in isolation [4]. Quantitative CT has certain theoretical advantages as it distinguishes cortical from trabecular bone. Both, however, are limited to providing information on bone density and tell us little about bone quality.
Regulation of Bone Remodelling – A Basis for New Therapies?
How is the intricate balance between bone formation and resorption maintained? It has long been recognised that within the bone microenvironment, osteoblasts regulate the activity of neighbouring osteoclasts and, via specific receptors, coordinate the bone resorbing action of thyroxine, PTH and calcitriol, amongst others. While the paracrine release of cytokines (including interleukin-1, -6 and -11, TNF, TGF-β and M-CSF) ostensibly explained this cellular cross-talk, spatial proximity of osteoblast to osteoclast seemed to be a prerequisite for behavioural change in the latter. Growth factors in isolation provided an inadequate stimulus. The discovery of two glycoproteins, both from the TNF receptor superfamily, termed (after several early nomenclatural changes) osteoprotegerin (OPG) and the ligand for receptor activator of NF-ĸB (RANK-L) established the missing link. These constitute a system that acts as the final effector pathway for a host of osteotropic stimuli. RANK-L expression is inducible on the osteoblast cell membrane, while its receptor (RANK) is found on cells of an osteoclastic lineage. Binding of ligand to receptor promotes osteoclastic differentiation, activation and survival, thereby increasing the pool of mature osteoclasts available to undertake bone resorption. OPG provides an ingenious regulatory mechanism. Like RANK-L it is an osteoblastic product, but is secreted rather than membrane bound. Once released, it acts as a decoy receptor for RANK-L, preventing its interaction with RANK and slowing the recruitment of osteoclasts. In this way, bone remodelling becomes intimately related to changes in expression of OPG and RANK-L. Factors that decrease the OPG/RANK-L ratio, thereby favouring bone resorption with higher rates of bone turnover, include TNFα, PTH, calcitriol, glucocorticoids and cyclosporine. Those that increase the ratio, thereby decreasing bone resorption and slowing bone turnover, include oestrogens, androgens and, possibly, IGF-I. In transgenic mice, OPG over-expression, or RANK-L or RANK knockout, all result in osteopetrosis, while OPG knockout causes severe osteoporosis [5,6,7]. This latter finding, coupled with the observation that OPG administration prevents bone loss in ovariectomised mice, has stimulated interest in the development of OPG as a treatment for osteoporosis. Encouraging clinical data are already emerging, including the demonstration of reduced bone turnover, inferred from the biochemical response, in post-menopausal women after a single OPG dose [8]. OPG could have potential for the prevention and treatment of post-transplant osteoporosis, where bone mineral loss is often profound and related to the uncoupling of bone resorption (calcineurin inhibitors and persistent hyperparathyroidism) and formation (corticosteroids). Measurement of serum OPG concentration also shows promise as a surrogate marker of bone turnover and, possibly, cardiovascular risk.
Parathyroid Hyperplasia – Still a One-Way Ticket
It is important to remember that, effects on bone apart, SHPT is defined in terms of three related, though pathophysiologically distinct, characteristics: (i) increased PTH synthesis per cell, (ii) hypersecretion of pre-synthesised PTH and (iii) progressive parathyroid hyperplasia. The ability of calcitriol to abrogate the first of these, through the powerful suppression of PTH gene transcription, has secured active vitamin D a central role in day-to-day clinical management. However, its ability to influence the last, parathyroid hyperplasia, is more limited. Early on, both directly and through increasing serum calcium concentrations, calcitriol can inhibit cell proliferation, though this can be overridden by concomitant hypocalcaemia and/or hyperphosphataemia. Clonal proliferation of parathyroid cells supervenes and is associated with reduced expression of both vitamin D and calcium-sensing receptors (CaR), restricting inhibitory inputs on two fronts. Irreversible nodular hyperplasia develops, and SHPT becomes refractory to treatment. Although calcimimetic agents (see later) have been shown to prevent parathyroid proliferation in the uraemic rat [9], their effect in long-term human disease remains to be seen. For the moment, significant hyperplasia remains a one-way ticket to parathyroidectomy.
When to Start Treatment?
The pathogenesis outlined above suggests that earlier intervention and prevention of SHPT should be more prominent therapeutic goals. In the pre-dialysis setting, overt increases in serum phosphorus are a relatively late development and PTH is a better marker for derangements necessitating treatment. Conventionally, increases in PTH have triggered the prescription of calcium-based phosphate binders to supplement calcium levels and prevent phosphorus elevation, with alfacalcidol or calcitriol added when the PTH response is deemed inadequate. Whilst such vitamin D analogues might be effective in suppressing PTH production and parathyroid cell growth in this setting, the increased risk of adynamic bone disorder means that there may be a significant price to pay. Vitamin D insufficiency, as manifest by a low plasma 25-hydroxyvitamin D [25(OH)D] concentration, is itself a relatively common finding in the chronic kidney disease (CKD) population beyond stage 3 (glomerular filtration rate <60 ml/min) and may be associated with early reductions in bone mineral density and SHPT. In this context, replacement with ergocalciferol or cholecalciferol seems reasonable, though remains of unproven benefit [10].
Achieving Therapeutic Targets
Target concentrations for PTH, phosphorus and calcium in the various stages of CKD have recently been outlined in a substantial document produced by the National Kidney Foundation Kidney Disease Outcomes Quality Initiative (table 1) [10]. Suggested phosphorus levels for patients with advanced CKD (stages 3–5) are close to the normal physiological range and may prove difficult to achieve in practice. The target level of corrected total calcium is quite low and very challenging at 2.10–2.37 mmol/l. In the case of PTH, the target concentration is close to normal in patients with CKD up to stage 3. In CKD stages 4 or 5, however, the target PTH level is higher (3–5 times the upper limit of normal), reflecting a body of evidence suggesting that moderate elevations in PTH are more likely to be associated with normal bone turnover. The practising clinician will immediately recognise the difficulties that these targets present to existing therapies; a recent analysis of a large cohort of Spanish patients showed just 13% at target for both PTH and Ca × P and 22% for PTH alone [11]. Available best therapy is clearly struggling to deliver the results now demanded.

Calcimimetics
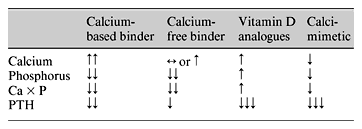
The CaR is widely expressed, but it is in parathyroid and renal tissue that its function is best understood. As the physiological role of the CaR has been unravelled, expectation has grown that its pharmacological manipulation will provide the means to achieve and maintain a target PTH. Cinacalcet (formerly AMG 073) and NPS R-568 are allosteric modulators of the CaR, enhancing its sensitivity to ambient calcium and, thereby, countering a key pathophysiological development – under-expression of the parathyroid CaR and partial loss of calcium-driven PTH suppression. The efficacy of cinacalcet has been demonstrated in a number of clinical trials, with impressive reductions in PTH accompanied by declines in calcium, phosphorus and Ca × P (table 2) [12,13,14]. These latter observations are thought to reflect a reduced efflux of calcium and phosphorus from bone as PTH falls, though functioning CaRs within the skeleton could potentially play a role. Consequently, respective roles of calcitriol and cinacalcet as therapy for SHPT are likely to be determined, not solely by the serum concentration of PTH, but also by the prevailing calcium, phosphate and Ca × P. In circumstances where calcium and Ca × P are low, treatment would typically start with calcitriol. Cinacalcet would be favoured as initial therapy when calcium or Ca × P are high. Calcimimetic agents should enable more precise control of serum PTH concentration than previously possible and help us, not only to achieve the appreciably more stringent treatment targets now recommended, but hopefully also to limit both skeletal and cardiovascular morbidity in patients with CKD.
Vitamin D Analogues
In the experimental setting, the newer generation of vitamin D analogues have shown considerable therapeutic potential. This is especially the case for 22-oxacalcitriol (licensed in Japan) and paricalcitol (licensed in the USA). Unfortunately, under the clinical spotlight none of these compounds has been shown to be truly non-calcaemic and such descriptions of them remain misleading. The majority of available studies are placebo-controlled only and highly reminiscent of reports heralding calcitriol and alfacalcidol in the 1970s. Comparative studies are rare, although paricalcitol offered a marginal advantage over calcitriol with respect to sustained elevations of calcium and Ca × P product in a head-to-head study [15]. Very recently, a large cohort study suggested a possible survival advantage for those patients treated with paricalcitol [16]. Although perhaps counterintuitive, the results of this analysis are potentially very important, serving to emphasize the need for large, randomised, prospective studies comparing newer analogues with more established compounds. At present, there remains insufficient evidence to justify a wholesale shift toward the newer vitamin D agents. Nevertheless, the future development of ‘designer’ vitamin D compounds, whose distinct pharmacological characteristics afford them an edge in the clinical arena, remains an appealing prospect.
New Phosphate Binders
Hyperphosphataemia directly stimulates PTH synthesis, accelerates parathyroid hyperplasia and forecasts a poor response to treatment with vitamin D. Its significance is not limited to the parathyroid/skeletal axis, however – hyperphosphataemia predicts cardiovascular mortality in dialysis patients and, as part of the Ca × P, is a risk factor for soft tissue and vascular calcification. Moreover, phosphate appears to be more than just a passive participant in the process of uraemic vascular calcification – its ability to accelerate the transformation of vascular smooth muscle cells to an osteoblastic phenotype, capable of in vitro bone matrix production, pointing to a more proactive role (see below) [17].
It is important to remember that the control of serum phosphate is not the exclusive responsibility of phosphate binders. The contribution of the dialysis process to achieving normophosphataemia is just as important. The complex nature of phosphate handling on haemodialysis mandates the need for regular, extended sessions for adequate removal – the compromise of thrice weekly schedules represents the bare minimum. Conversion from conventional prescriptions to daily nocturnal haemodialysis virtually eliminates phosphate binder requirements [18].
The ideal phosphate binder would safely and effectively lower serum phosphate when used in a single, preferably once-daily, regimen whilst not engendering any significant clinical drawbacks. Aluminium hydroxide is reasonably effective but highly toxic in some patients, while an elevated calcium burden has cast a cloud over calcium-based binders, particularly in the setting of concurrent vitamin D administration. Sevelamer hydrochloride is at the vanguard of a new generation of calcium and aluminium free phosphate binding agents with others, including lanthanum carbonate, in the pipeline. Considered in terms of ability to reduce serum phosphate, sevelamer is comparable to, but no better than, conventional binders. Thus, although Ca × P product was a primary end point for the ‘Treat to Goal’ study, no significant differences were found between sevelamer and calcium-containing binders [19], and a calcium acetate vs. sevelamer study suggested that calcium acetate is superior to sevelamer in maintaining Ca × P product below a 55-mg2/dl2 target [20]. Sevelamer certainly has potentially important advantages in that it does not increase calcium load and is associated with fewer hypercalcaemic episodes than calcium-based agents. It has been shown to reduce the tendency to soft tissue calcification and renal damage in the uraemic rat [21] and attenuate the progression of coronary and aortic calcification in haemodialysis patients [19]. It also has the added bonus of reducing total and LDL, while increasing HDL, cholesterol. It is generally well tolerated, though gastrointestinal side effects are not infrequent. It is, however, extremely expensive, a fact likely to preclude its use in a number of countries.
The Rise and Rise of ABD
The historical link between ABD and aluminium toxicity has little contemporary relevance. The major present-day contributors to ABD are treatment with active vitamin D, calcium overload and choice of peritoneal dialysis as modality of renal replacement therapy. In addition, in a significant proportion of the CKD population, the coexistence of other threats to the skeleton, including age-related, or post-menopausal osteoporosis and diabetes mellitus may predispose to the adynamic lesion.
A low serum PTH, often in combination with hypercalcaemia, is our clinical surrogate for ABD. Though useful, this may serve to exaggerate the importance of PTH in the evolution of the disorder. The occurrence of ABD in those with persistent elevations of PTH implies a degree of PTH independence. Here, active vitamin D therapy such as calcitriol may be culpable through an inhibitory influence on osteoblast function. Support for this comes from the differential properties, at the level of bone, of the new analogues of vitamin D. For example, these have demonstrable differences in their ability to stimulate the production of IL-6, an important paracrine mediator of bone cell function [22]. In addition, the analogue 22-oxacalcitriol has been shown to suppress PTH in the uraemic dog without increasing the risk of adynamic bone [23]. PTH and calcitriol orchestrate the later steps in osteoblast and osteoclast differentiation. Other local growth factors and cytokines coordinate the early stages; anomalies in the synthesis and secretion of these may be more relevant to the development of ABD. Moreover, since the classic osteotropic hormones and local factors operate through the RANK-L/OPG system, this pathway merits intense scrutiny in the context of the pathophysiology, diagnosis and treatment of ABD.
At the Interface of Bone and Vascular Biology
The recent emergence of electron beam CT and electrocardiogram gated multi-slice CT as viable techniques for the non-invasive imaging of coronary vessels and the aorta has rekindled interest in the contribution of vascular calcification to the excess cardiovascular mortality encountered in the dialysis population. Moreover, the finding of a positive association between coronary artery calcification and oral calcium intake has called into question the future role of both calcium-containing phosphate binders and calcaemic vitamin D analogues. The discovery of spontaneous arterial calcification in mice carrying specific gene deletions coupled with the recognition of certain hitherto ‘bone-specific’ regulatory proteins in calcified arteries, and the ability of several stimuli (including inorganic phosphate [17] and uraemic serum [24]) to provoke calcification in cultures of vascular smooth muscle cells, has generated a new biological paradigm that departs from the traditional view of high calcium loads and elevations of the Ca × P product promoting soft tissue mineral deposition via a passive process only. The realisation that calcification is a complex, coordinated process, with an intimate relationship to skeletal metabolism has inevitably led to a re-examination of the relative contributions of hyperphosphataemia, high Ca × P product, SHPT, ABD, uraemia per se and currently available treatments to its development and progression. It also raises the prospect of future ‘crossover’ treatments; for example bone morphogenetic protein BMP-7 has recently been shown to have beneficial effects in murine models of both uraemic vascular calcification [25] and SHPT [26], while OPG, a promising treatment for bone loss, has also been shown to inhibit vitamin D-induced vascular calcification [27]. This integration of bone and vascular biology cannot fail to influence the way we perceive and effect the treatment of renal and other bone diseases in the future.
Conclusion
A rapid expansion in our understanding of the bone remodelling process and its relationship with vascular calcification has emphasised the complex, heterogeneous nature of renal osteodystrophy and led to its emergence as a potentially modifiable cardiovascular risk factor. This is reflected in new, more stringent, therapeutic targets – targets that, while perhaps out of the reach of conventional treatments, may prove more realistic as novel agents are developed. Although hyperphosphataemia remains a major problem, with recent developments in phosphate control at the margin only, it seems inconceivable that our day-to-day practice with respect to the diagnosis and management of the skeletal complications of CKD will not change significantly in the near future.
