

Abstract
Background/Aims: Proteinuria, nearly a universal finding in progressive kidney disease, has been the subject of frequent recent analyses in the renal literature. Proteinuria is a hallmark of diabetic nephropathy: microalbuminuria is the principal early predictor for progression of diabetic glomerulopathy, and proteinuria may be viewed as a measure of the severity and promoter of progression of nephropathy. Methods: This article critically reviews for the first time the full scope of diabetic proteinuria – complex molecular mechanisms, natural history, and analysis of treatment trials – in order to address the validity of ‘the proteinuria hypothesis’, i.e., that diabetic proteinuria is a modifiable determinant of renal progression. This hypothesis is analyzed in detail, including recent studies on the primary therapy of diabetic nephropathy, renin-angiotensin blockade. Results: As fully developed, this hypothesis consists of three postulates: that higher amounts of proteinuria predict progressive loss of function, that proteinuria reduction correlates with slowing progression, and that proteinuria is a surrogate endpoint for clinical trials. The latter postulate has not before been adequately linked to growing information about the first two postulates as they apply to diabetic kidney disease. Conclusion: While diabetic nephropathy is a disease model for the potential use of proteinuria as a surrogate marker for renal progression, this shift in perspective will require prospective data from additional clinical trials, particularly of non-renin-angiotensin blocking drugs, to be complete.
Introduction
Diabetic kidney disease is characterized by excessive urinary albumin excretion followed by loss of kidney function. Proteinuria is a hallmark of diabetic nephropathy. The number of Americans diagnosed with diabetes mellitus has increased 61% over the last decade and will more than double by 2010. Data from five consecutive cross-sectional US national surveys, most recently NHANES 1999–2000, indicate that the most dramatic increase in diabetic cases has been in the most obese individuals [1], where the prevalence in 2000 was 3 times the level in 1960. Amidst an epidemic of diabetes, the disease has become the most common single cause of chronic kidney disease in the US and Europe [2, 3]. The incidence of diabetic nephropathy has more than doubled in the past decade [4], due largely to increasing prevalence of type 2 diabetes [4]. Microalbuminuria was present in 32.8% of adults with previously diagnosed diabetes in NHANES III (1988–1994) and in 27.5% of adults in NHANES 1999–2000 [5]. Diabetic nephropathy now accounts for about 40% of new cases of end-stage renal disease (ESRD) [6]. A recent study has estimated the annual health care costs of diabetic nephropathy in the US at USD 1.9 billion for type 1 and USD 15.0 billion for type 2 diabetes [7]. Nearly a universal finding in progressive kidney disease [8], proteinuria, predominantly albuminuria, has long been accepted as the clinical hallmark of diabetic nephropathy [9], and its most common laboratory manifestation (fig. 1) [10]. In NHANES II, 1% of the general adult population and 6.1% of diabetics screened demonstrated macroalbuminuria [11].
![Fig. 1. Proteinuria (a) and progression to ESRD (b) in diabetic nephropathy in type 1 and type 2 diabetic patients. Similar rates of proteinuria and time of progression from onset of proteinuria to kidney failure occur in both types of diabetes [10].](https://karger.silverchair-cdn.com/karger/content_public/journal/ajn/25/2/10.1159_000084286/2/m_000084286_f01.gif?Expires=1716291236&Signature=MwFpi2vfoF6ueyInwAb7VPbyYRbmBlMgao1qabIIbd764HuTft8pF9-tjYBxw0tLQzaus9IzKlVNoqxkR7HCWmd9HxLWNl6QLjXmZNa-gR0XKzdx6-z-sgN4ayB5OEQf4MYxgAAIRodNbFRhUV25DjhumUOeQIB5qSWCdhimLVFj3BoIKamKW6tCfqgVj2N7J9zS54vsu8-BFIIP9Rv8Yfa7wGVaIuwNLF5rpD3n-nEFbs1jb5BmPgKdU92PbBTWsVVflQ4BYj5wQ-2Hl8mrXY3Ean3~YxCn4GHnTEpvOc3CHIZCPrbHyvtGvJa-39QkKRp2E7jv5I8AWV0-DVQeHA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Proteinuria (a) and progression to ESRD (b) in diabetic nephropathy in type 1 and type 2 diabetic patients. Similar rates of proteinuria and time of progression from onset of proteinuria to kidney failure occur in both types of diabetes [10].
Proteinuria (a) and progression to ESRD (b) in diabetic nephropathy in type 1 and type 2 diabetic patients. Similar rates of proteinuria and time of progression from onset of proteinuria to kidney failure occur in both types of diabetes [10].
This article reports on the complexity of diabetic proteinuria and of its primary therapy, renin-angiotensin blockade. It analyzes the current importance given to diabetic nephropathy in support of the ‘proteinuria hypothesis’, i.e., that diabetic proteinuria is a modifiable determinant of renal progression. The concept that reducing proteinuria is an effective way to halt progression of the disease [12] is evaluated in detail.
Overt diabetic nephropathy is characterized by persistent proteinuria (>0.5 g/24 h) or macroalbuminuria (>300 mg/24 h) [13]. In the natural history of the disease [14], proteinuria is preceded by stages of excessive glomerular filtration and of microalbuminuria, which signals an increased risk of progression to overt nephropathy. A progressive increase in proteinuria subsequently leads to a variable decline in renal function. While initial microalbuminuria may enter into remission [15], and fewer than half of microalbuminuric patients progress to higher levels of proteinuria [16], it remains the principal early predictor for progression at this time.
It is now widely accepted that proteinuria reduction is an appropriate therapeutic goal in chronic kidney disease with proteinuria [17]. Experimental and clinical studies continue to examine the role of proteinuria in diabetic nephropathy. Proteinuria signifies evidence of glomerular damage, and may be viewed as a measure of the severity of diabetic glomerulopathy. Heavy proteinuria in diabetic nephropathy is strongly associated with pathological changes of diffuse and, less commonly, the nodular form of diabetic glomerulosclerosis [18]. Early clinical reports noted nephrotic syndrome in 87% of type 1 and 70% of type 2 diabetic patients with nephropathy, and end-stage renal failure occurs in up to 75% of diabetic patients within 15 years of developing overt proteinuria [19]. The overall 10-year incidence of gross proteinuria in diabetic patients is about 33%, with a prevalence similar in type 1 and type 2 patients [14]. Rising urinary protein excretion confers increased risk of overall mortality and fatal cardiovascular events in patients with diabetes mellitus. Four decades after the diagnosis of diabetes, almost 3/4 of those lacking proteinuria are alive, in contrast to 1/10 of those with proteinuria [20].
Factors which cause progression of kidney disease continue to be actively investigated, and include glomerular hypertension and hypertrophy, activation of coagulation pathways, and lipid deposition. Proteinuria is commonly viewed as a nonhemodynamic promoter of disease progression in diabetic nephropathy [21, 22]. Treatment to delay progression of chronic renal failure now includes strict control of proteinuria [23] as the basis of therapy. Reducing proteinuria to less than 1 g/24 h has been added to the targets of glycemic control and lowered blood pressure goals in preventing progression. Two decades of progress in retarding the progression of renal disease were recently reviewed [24].
More recent attention has focused on the prevalent ‘proteinuria hypothesis’, i.e. that proteinuria is a target of new therapies for tertiary prevention in diabetic nephropathy [25, 26]. The proteinuria hypothesis consists of three postulates: (1) higher levels of proteinuria predict adverse clinical outcomes, (2) reduction in proteinuria correlates with slowing of renal progression, and (3) proteinuria is a surrogate endpoint and target of clinical trial interventions, in this case, for diabetic nephropathy. According to this hypothesis, measurement of proteinuria is correctly used to establish not only the diagnosis of overt diabetic nephropathy, but its risk of subsequent loss of renal function.
There is evidence to support the notion that the rate of progression of diabetic nephropathy may be best slowed or even reversed in its earlier stages [27]. However, clinical interventional trials for regulatory drug approval have historically targeted doubling of serum creatinine levels or patient mortality rates as primary outcome measures. Both measures are more likely to occur in later stages of diabetic nephropathy. Reducing proteinuria has emerged, then, as a potential outcome measure suitable for intervention in earlier stages of the disease. Proteinuria has been proposed as a surrogate which would allow shorter interventional trials involving nephroprotective therapy for diabetic nephropathy.
Mechanism
Understanding of the molecular mechanisms and ultrastructural changes causing proteinuria has made remarkable advances over the past few years [28], adding understanding to the previous concepts of glomerular barrier function [29]. While a full discussion of the mechanisms of the stage of microalbuminuria is beyond the scope of this review, it should be noted that the appearance of microalbuminuria is currently thought to be a consequence of generalized endothelial injury in the vasculature. Pathogenetic mechanisms involving the vasculature are likely to occur in glomerular endothelial cells and podocytes as well [30]. Several proposed mechanisms were reviewed by Garg and Bakris [31] to explain local (renal) and systemic vascular injury in diabetic patients with macroalbuminuria. MA is a powerful independent risk factor of cardiovascular disease in diabetes mellitus. Local injury to vascular smooth muscle cells and endothelial cells may increase vascular permeability in the kidneys and systemically. While systemic hypertension is the most important single determinant of MA in the diabetic, this relationship is more pronounced in patients with elevated C-reactive protein levels [32. ]MA is a strong predictor of total and cardiovascular mortality in both type 1 and type 2 diabetes [31].
Determining the mechanisms of proteinuria specific to diabetes has importance for each postulate of the proteinuria hypothesis. Of four mechanisms cited for excessive protein excretion in general – increased glomerular filtration, inadequate tubular absorption, overflow, and increased tubular secretion – the dominant mechanism is glomerular. Proteinuria primarily reflects glomerular injury and increased glomerular permeability to macromolecules. The exact molecular mechanisms causing the breakdown of the glomerular filtration barrier in diabetes remain to be fully defined. It is widely accepted that the passage of proteins across the filtration barrier can be affected by several factors, including the hemodynamic pressure gradient across the glomerular basement membrane and factors intrinsic to the filtration barrier, including the pore size and extent of anion charges [29]. While mechanisms altering the protein-selective properties of the diabetic glomerular capillary wall are not fully understood, evidence exists for hemodynamic as well as intrinsic basement membrane changes as causes of diabetic proteinuria (fig. 2) [33]. Angiotensin exerts complex hemodynamic and nonhemodynamic actions that may contribute to diabetic nephropathy, including induction of systemic vasoconstriction, increased glomerular arteriolar resistance, increase in glomerular capillary pressure, increased glomerular capillary permeability, reduction in the filtration surface area, stimulation of extracellular matrix proteins, and stimulation of renal proliferation and fibrogenic chemokines (table 1) [34,35,36]. The structural basis for the leak may reside within the glomerular basement membrane as well as in the nearby epithelial cell layer; specific mechanisms may include basement membrane damage due to lucent deposits, and focal foot process degeneration of the podocytes.

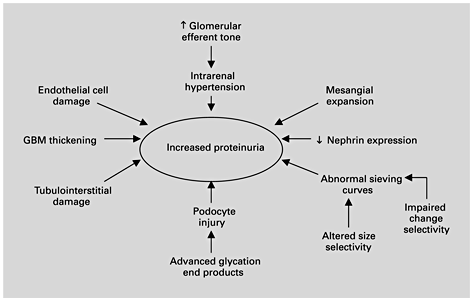
Mechanisms of proteinuria in diabetic nephropathy. GBM = Glomerular basement membrane.
Mechanisms of proteinuria in diabetic nephropathy. GBM = Glomerular basement membrane.
Several pathological changes are characteristic of diabetic glomerulosclerosis (fig. 3a), including nodular and/or diffuse sclerosis of glomeruli, increased basement membrane thickening, increased mesangial matrix, hyaline arteriolopathy of afferent and efferent vasculature, and varying degrees of interstitial fibrosis with tubular atrophy [4, 37]. Some renal structural abnormalities are known to precede the development of proteinuria in diabetic nephropathy, including increased basement membrane width and mesangial expansion [38]. The degree of both mesangial and interstitial expansion has been found to correlate with the degree of albuminuria. Progressive mesangial expansion may be due to glycoproteins, collagen, or other factors [39]. Electron-dense deposits in or around the glomerular basement membrane may restrict the glomerular capillary filtration surface area and lead to proteinuria.
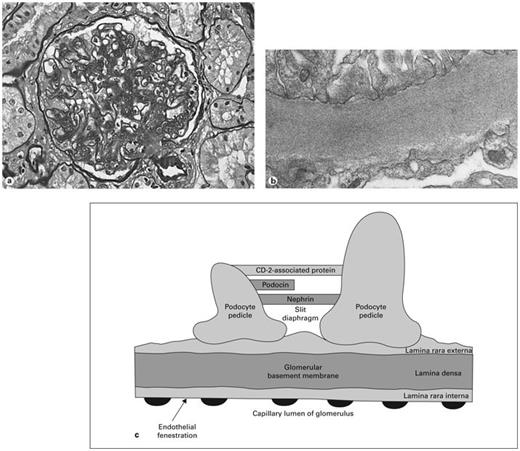
a Pathological changes of advanced diabetic nephropathy include increased glomerular basement membrane thickness, mesangial matrix expansion, nodular glomerulosclerosis, glomerular hypertrophy, and arteriolopathy of afferent and efferent arterioles. b Electron microscopy showing increased basement membrane thickness. c The barrier to proteinuria. Schematic drawing of the visual glomerular epithelial cells (podocytes) lining the outer aspect of the glomerular basement membrane. Foot processes are connected by the slit diaphragm with nephrin, podocin, and other proteins. Proposed mechanisms of diabetic proteinuria include structural changes to the basement membrane, hemodynamic injury to podocytes, decreased number of podocytes, damaged slit diaphragm components, and reduced expression of nephrin.
a Pathological changes of advanced diabetic nephropathy include increased glomerular basement membrane thickness, mesangial matrix expansion, nodular glomerulosclerosis, glomerular hypertrophy, and arteriolopathy of afferent and efferent arterioles. b Electron microscopy showing increased basement membrane thickness. c The barrier to proteinuria. Schematic drawing of the visual glomerular epithelial cells (podocytes) lining the outer aspect of the glomerular basement membrane. Foot processes are connected by the slit diaphragm with nephrin, podocin, and other proteins. Proposed mechanisms of diabetic proteinuria include structural changes to the basement membrane, hemodynamic injury to podocytes, decreased number of podocytes, damaged slit diaphragm components, and reduced expression of nephrin.
Glomerular permselectivity (fig. 3b) involves two adjacent molecular filters: the glomerular basement membrane and the slit diaphragm [40]. The glomerular basement membrane in humans (fig. 3c) is a complex three-layered structure comprised of endothelial cells with fenestrations, the dense fibrillar glomerular basement membrane itself, and podocytes, the outer visceral epithelial cells. (Previously viewed as an insignificant part of the barrier, recent data suggest that the endothelium may play a role in glomerular permselectivity [41]. The endothelial barrier properties may derive from its cell coat, or glycocalyx.) Between the interdigitating foot processes of the podocytes arises the slit diaphragm, a zipper-like complex. In humans the basement membrane layer, attached to endothelial and visceral epithelial cells, has a thickness of 250–350 nm. The most abundant single component of the filtration membrane is type IV collagen.
The progression from normal albumin excretion to overt diabetic proteinuria correlates with the loss of size and charge selectivity of the filtration barrier [18]. Dysfunction of the size selectivity properties of the intrinsic glomerular barrier to plasma protein filtration has been evaluated in both type 1 and type 2 diabetes. Fractional clearances of albumin, IgG, and neutral dextrans suggest a distribution of glomerular basement membrane pore radius toward larger pore sizes [18, 42]. Concomitant changes in charge selectivity as well may contribute to albuminuria in type 2 diabetes. In other experimental models of diabetic nephropathy, proteinuria is associated with a reduction in slit pore density.
Renal hemodynamics play a critical role in renal hyperfiltration of early diabetes, where glomerular filtration rates are supranormal [43]. A dominant theory based on animal models of diabetic nephropathy has been that glomerular hypertension secondary to reduced intrarenal vascular resistance leads to glomerular structural damage, including endothelial and epithelial cell damage, and disruption of the filtration barrier [44]. More recent investigation has focused on the roles of podocyte injury and nephrin in diabetic nephropathy. One of the most prominent ultrastructural abnormalities is podocyte loss [45]. Injury and loss of podocytes is a prominent ultrastructural abnormality in diabetic nephropathy [46, 47]. Visceral epithelial cell injury and reduction in the number of podocytes per glomerulus leave fewer podocytes to cover the surface area [48]. Increased synthesis of collagen by podocytes could lead to glomerular basement membrane thickening, or excessive amounts of secreted vascular endothelial growth factor could enhance permeability of the barrier to macromolecules. Several mechanisms of podocyte loss have been speculated. Mutations in podocyte genes have also been implicated in susceptibility to glomerular disease. A recent study reported that loss of CD-2-associated protein, a component of the glomerular filtration complex, may modulate disease not only because of its structural role in the slit diaphragm, but because it also directs proteins to the podocyte degradative pathway [49].
Nephrin is a 1,241-residue transmembrane cytoskeleton protein gene product localized to the filtration slit area between foot processes of the podocytes, and is essential for the formation of the zipper-like slit diaphragm structure [50]. Adler [51 ]emphasized the complexity of diabetic proteinuria in a recent Nephrology Forum review of podocyte pathology and nephrin in diabetic nephropathy. The author argued that podocyte injury in diabetic proteinuria may be mediated by signal transduction changes, cytoskeletal injury, alterations in the podocyte slit pore membrane, detachment from the glomerular basement membrane, and apoptosis. Adequate experimental data support the above mechanisms of podocyte injury. However, changes in the slit diaphragm related to nephrin expression, also reviewed by Adler, have been far more extensively explored. Nephrin expression is regulated in a complex way [50], and reduced gene production or dislocation of nephrin has been implicated in nondiabetic models of proteinuria kidney disease [52, 53]. A reduction in nephrin expression may be a determinant of glomerular protein leakage in experimental diabetic nephropathy [54,55,56]. Ultrastructural and immunohistochemical studies have shown that nephrin is diminished in the glomeruli of diabetic rats [51, 55, 57]. In the diabetic rat model, reduction in glomerular nephrin gene expression determined by quantitative real-time PCR is temporally associated with increasing albuminuria [55, 58]. Some human data also suggest a downregulation of nephrin expression in the diabetic kidney, in both type 1 and type 2 diabetic nephropathy, perhaps mediated by angiotensin II and glycated albumin [59]. A recent study by Langham et al. [60 ]reported a 62% reduction in nephrin expression in glomeruli from patients with diabetic nephropathy. Expression of nephrin mRNA was reduced and inversely related to the severity of proteinuria in another recent small study of type 2 patients with nephropathy [61]. A gene expression profiling study using oligonucleotide microarray analyses reported the nephrin gene to be among those downregulated in diabetic nephropathy [62]. Reduced nephrin mRNA and protein expression may be associated with podocyte ultrastructural abnormalities, providing one potential mechanism for proteinuria in diabetic nephropathy [63, 64]. A potentially important interaction linking angiotensin II and nephrin was also reviewed in the analysis by Adler [51]. Nephrin expression decreases acutely when podocytes in culture are incubated with angiotensin II, suggesting a direct influence of angiotensin on nephrin. Recent experimental data have shown that renin-angiotensin system (RAS) blockade results in a change in nephrin expression in diabetes [54, 58]. Furthermore, nephrin mRNA expression, reduced in diabetes, can be preserved by angiotensin-converting enzyme inhibitor (ACEI) [60]. The signaling pathway between angiotensin and nephrin expression is not clear. These reports suggest that the RAS affects proteinuria in diabetic nephropathy in part by effects on nephrin, and that ACE inhibition might modulate proteinuria in diabetic nephropathy by ameliorating diabetes-induced changes in nephrin synthesis.
The K/DOQI Work Group has recommended that urinary albumin excretion is the appropriate measure for monitoring chronic kidney disease [65] and that albumin and total protein excretion are correlated. Recently it has been proposed that the concept of increased albumin permeability as the sole abnormality causing proteinuria, with no defect in tubular retrieval of albumin, is inadequate [66]. Albumin and other plasma proteins filtered through the glomerulus undergo tubular absorption and degradation. As a result, albuminuria may be importantly influenced by tubular albumin metabolism. The fragmentation of filtered albumin is extensive, so that albumin in the urine may exist in conventional immunoreactive as well as conventionally unmeasured, immunounreactive forms [67]. A recent study proposed that in twenty diabetic patients with macroalbuminuria, large quantities of protein fragments not routinely measured clinically were present [68]. The role of defects in albumin retrieval in progression of diabetic nephropathy deserves further exploration.
It has long been understood that, as loss of kidney function progresses in diabetes, tubulointerstitial dysfunction in the form of low-molecular-weight tubular proteinuria may also become evident [69]. Diabetic nephropathy is now understood to be a disease which affects the entire nephron, i.e. the tubulointerstitium as well as the glomerulus [70]. Severity of proteinuria has been correlated with the extent of tubulointerstitial injury in experimental nondiabetic renal disease [71, 72]. Tubulointerstitial injury has been the focus of increasing investigation in relation to glomerular injury and as an important pathological predictor of kidney impairment in diabetes [73]. Pathology of the interstitium in diabetic nephropathy includes tubular basement membrane thickening, tubular fibrosis and atrophy, and arteriolosclerosis. Interstitial expansion correlates with the extent of albuminuria, and interstitial fibrosis correlates more closely with loss of function than does glomerular injury [70].
Not specific to diabetes, a more recently developed corollary of relevance to the proteinuria hypothesis is that increased protein filtration and excretion may contribute to the pathogenesis of glomerular lesions and disease progression in nephropathy [74, 75]. The proposed effects of proteinuria on the kidney include increased severity of glomerulosclerosis as well as tubulointerstitial injury, in both experimental models of proteinuric kidney disease and in humans. The degree of glomerular proteinuria is known to correlate with histologic interstitial fibrosis [76]. Filtered and reabsorbed proteins cause release of vasoactive and inflammatory substances into the interstitium of the kidney, resulting in fibroblast proliferation and interstitial inflammation. Mechanisms which have been proposed for the dangers of proteinuria include direct mesangial toxicity, toxicity of specific filtered proteins, and induction of proinflammatory molecules [77, 78]. More recent data also suggest that proteinuria is responsible for tubular cell injury as well as interstitial damage, due to tubular overload of proteins, exceeding the lysosomal capacity for endocytosis [79]. The complex reaction includes lysosomal rupture and release of endothelial and mononuclear cell chemotactic proteins. Excessive filtered protein reaching the proximal tubule may lead to peritubular inflammation and fibrosis. Recently reported mechanisms of tubulointerstitial injury involving the proximal tubule attributed to proteinuria include regulation of proximal tubular gene expression profiling [80] and modulation of chemokine production [81, 82] by proteinuria. These complex and multifactorial mechanisms linking proteinuria and tubulointersitial injury give further support to the first postulate of the proteinuria hypothesis.
Natural History
Clinical proteinuria was first noted in patients with diabetes over a century ago, and was described in association with diabetic glomerulosclerosis in a pathological report by Kimmelstiel and Wilson [83 ]in 1936. The sequence of proteinuria followed by loss of renal function was not described until 40 years later. Only a decade later was another distinct stage of nephropathy, microalbuminuria, identified, in type 1 diabetes. These features of the natural history of diabetic nephropathy are now widely known (fig. 4).

Natural history of diabetic nephropathy, including changes in glomerular filtration and proteinuria over time. Proteinuria reduction is shown as a tertiary prevention. GFR = Glomerular filtration rate.
Natural history of diabetic nephropathy, including changes in glomerular filtration and proteinuria over time. Proteinuria reduction is shown as a tertiary prevention. GFR = Glomerular filtration rate.
Diabetic nephropathy is clinically defined by the presence of persistent proteinuria greater than 0.5 g/day. The albumin excretion rate in patients with clinical proteinuria is generally >200–300 µg/min [84]. The average time to proteinuria from the time of diagnosis of type 1 diabetes is 19 years. Proteinuria tends to increase over time in untreated patients, and the majority of such patients become nephrotic. The yearly increase in protein excretion averages about 20%, but wide variation occurs in both incipient (microalbuminuria) and overt nephropathy [85]. Over a 2-year period in one study, average urine protein excretion increased from 1.8 to 3.3 g/day [86, 87]. The amount of total proteinuria in diabetic nephropathy patients is typically subnephrotic (<3.5 g/24 h) or nephrotic, with as many as 70–80% being nephrotic in some earlier studies of type 1 and type 2 patients with nephropathy [88]. Hypertension is an early feature in the course of persistent proteinuria, even when renal function is normal or only slightly impaired [89, 90]. Diabetic patients with persistent proteinuria have a high risk of developing proliferative retinopathy.
Without intervention, renal function falls over a period of several years once overt nephropathy is present, but at a highly variable rate. In many cases, diabetic patients with heavy proteinuria have the worst renal prognosis. Proteinuria may decline as glomerular filtration falls and renal failure becomes advanced. In type 1 patients with clinical proteinuria, glomerular function declines by about 12 ml/min/year. ESRD develops in 50% of patients over 10 years and in 75% over 20 years. In type 1 patients, on average, the natural history of diabetic nephropathy leads to death 6 years after the start of persistent proteinuria, although the range appears to be very wide. The overall sequence is similar in type 2 patients, but uncertainties may exist because of inaccurate dating of the onset of diabetes and a higher rate of nondiabetic or atypical features of diabetic glomerulosclerosis. The decline is more variable in type 2 patients [6] and the progression rate to ESRD may be as low as 20% over 20 years. However, the progression of renal failure, while constant in individual patients, varies widely among patients. Improvements in the management of nephropathy have extended the time course from persistent proteinuria to renal failure in comparison to these historical estimates.
Albumin, a small- to medium-sized molecule, is the protein that has been most extensively studied. As noted by Mogensen and Christensen [86] in 1984, the albumin excretion rate that corresponds to clinical proteinuria is generally greater than 200–300 µg/min. In patients with microalbuminuria, yearly increases in the albumin excretion rate of about 25 µg/min have been shown [90], although the range is wide [89]. Wide standard deviations are seen in the yearly increase in albumin excretion rate, but the vast majority of patients, particularly in type 1 diabetes, progress to clinical proteinuria over a few years. In its most advanced stages, diabetic glomerular proteinuria becomes less selective [91], with a significant leak of large proteins including both albumin and IgG, and tubular proteinuria leads to larger losses of markers such as β2-microglobulin [18, 69, 92]. While diabetic patients with heavy proteinuria exhibit an abnormal enhancement of very high molecular weight dextran clearance, the changes are small compared to a 1,000-fold increase or more in albumin excretion [84]. Newer parameters suggested to monitor the progression of diabetic nephropathy include the measurement of immunoglobulin subclasses, such as IgG4, which may be differentially eliminated due to charge as well as to nonenzymatic glycation [93].
Proteinuria Predicts
The first postulate of the proteinuria hypothesis is that proteinuria is an independent predictor of ESRD, so that the level of measured protein losses is an explanatory variable for the severity of glomerular damage. The postulate accepts the existence of other independent prognostic factors of renal progression, such as hypertension, genetics, or dietary intake.
More than 20 years ago, Hostetter et al. [94 ]proposed that proteinuria by itself could exacerbate glomerulosclerosis. A variety of experimental models and human kidney diseases have now indicated that baseline proteinuria should be accepted as an independent and modifiable risk factor for renal disease [95], at least certain levels of proteinuria in certain conditions. More severe proteinuria is correlated with a faster rate of progression to renal failure [72, 96, 97]. In the Modification of Diet in Renal Disease (MDRD) study (which excluded diabetic nephropathy patients), for example, baseline proteinuria was one of six factors and a continuous variable which independently predicted faster glomerular filtration rate decline [98]. Other studies have linked proteinuria to ESRD and renal death [97].
The effects of proteinuria on the kidney may be detrimental well under nephrotic levels of protein excretion; the adverse association of proteinuria with loss of function is stronger in patients with over 1 g of total protein excretion per 24 h [95, 99]. However, it should be recognized that a main limitation of proteinuria as a marker of chronic kidney disease is that the inherent intraindividual variation in urinary protein excretion is even greater than that for serum creatinine alone [100]. Similar to the rate of decline of renal function itself, excretion of total protein or albumin in the urine varies within an individual [87], up to a standard deviation of as much as 40–50%. It also appears that many exceptions occur to the link between proteinuria and poor outcomes [30, 101].
Evaluated in a diverse number of glomerular diseases, proteinuria has become a key clinical issue as a predictor of renal progression in human diabetic nephropathy, either at baseline levels or as a response to clinical intervention. Almost 40 years ago, Gellman et al. [102 ]reported a relationship between renal biopsy results of glomerulosclerosis and clinical proteinuria in type 1 diabetes. In 1972, Watkins et al. [9] reported that, while daily 24-hour proteinuria in diabetic nephropathy showed considerable variation, nephrotic proteinuria was associated with advanced histologic changes, and that the chief difference between those who survived and those who did not was heavy proteinuria [14]. In the landmark Collaborative Study Group trial of type 1 diabetic nephropathy, heavy proteinuria doubled the risk of nephropathy progression [22]. Patients who reached serum creatinine doubling endpoints had baseline total protein excretion of 4.99 g/day compare to 2.34 g in those who did not reach creatinine doubling. In the phase II clinical trial of the AGE inhibitor pimagedine in a similar population, subset analysis showed that those with the largest protein excretion were again at greatest risk for doubling of creatinine [103].
A decade ago, a study of 121 hypertensive type 2 diabetic patients with nephropathy revealed that loss of renal function was greater in patients with overt nephropathy at baseline (urine albumin excretion >300 mg/24 h) compared with patients enrolled with lower amounts of proteinuria [104]. Of two recent well-known studies on the use of angiotensin receptor blockers (ARBs) in patients with nephropathy due to type 2 diabetes, the Irbesartan Diabetic Nephropathy Trial (IDNT) [105] and the Reduction of Endpoints in NIDDM with the Angiotensin II Antagonist Losartan (RENAAL) trial [106,] proteinuria was a prospective outcome measure only in the latter. In the IDNT, baseline proteinuria averaged 2.9 g/24 h in the irbesartan, amlodipine, and placebo groups. No data were reported on the relation of baseline proteinuria to renal outcome measures [105]. Unpublished data revealing a 14× increase in risk of progression to advanced disease when baseline proteinuria equaled 3–4 g/24 h versus those with 1–2 g/24 h were included in a recent review by Weir [107 ]on diabetes and hypertension. In the RENAAL study, proteinuria was a secondary endpoint. Median urinary albumin/creatinine ratios were 1,237 in the losartan group and 1,261 in the placebo group. Urinary albumin excretion tended to correlate with stage of hypertension at baseline [108]. While no relationship of baseline proteinuria to renal outcomes was included in the original report, subsequent analysis reported proteinuria to be the most important predictor of ESRD [109]. A recent target presented further outcome analysis of the secondary endpoint of albuminuria from the RENAAL study [110] including the effect of the amount of albuminuria at baseline. Baseline albuminuria was directly related to renal outcomes, and was a stronger predictor than other well-known risk parameters such as serum creatinine, blood pressure, HbA1C, age, and ethnicity. The mean baseline albumin was 1.8 g/g of urine creatinine. When baseline albuminuria was divided into high (73.0 g/g), intermediate (71.5 < 3.0 g/g) and low (<1.5 g/g) subgroups, the renal endpoint was 5 times higher for the high versus the low subgroup (fig. 5) even when adjusted for baseline risk markers. Data on total protein excretion were not presented. This large clinical trial has provided the firmest data available on the importance of large quantities of albumin to renal progression in diabetic nephropathy.
![Fig. 5. Effect of baseline albuminuria on renal endpoint (combined endpoint of doubling of serum creatinine, ESRD, or death) and on the individual component of ESRD in the RENAAL study. Adjusted for baseline risk markers, the renal endpoint was 5-fold higher and risk of progression for ESRD 8-fold higher in the high versus the low albuminuria group [see 101].](https://karger.silverchair-cdn.com/karger/content_public/journal/ajn/25/2/10.1159_000084286/2/m_000084286_f05.gif?Expires=1716291236&Signature=m1r2k35fLZHctPB-wpVmLThO0dlOtKJwChhaFNPDrIGmsjUmODoWwHZd7M8xT5mcCpeUqmre-t6ObC0AsTgHFcgy-nIxKMonlnSr~XI5mEb-f4Wc6exQJ-e-Qzlh9xZ-qFDsk8nKyB8OSkTyNM7pvciM-epXYZ63QRh4eomRdShxjkUmB-fpEHS~VcsCWgodFmDTwv4JSW0nu8htF-xyBuQn~QLAdMded-Q3kZYG3FWLM~WntsbJySDx1~u6UzRsmOlWVx9LqmycGJLHSTo6-wcrQsVwXuxws~3dyn6SZiT8FGhFKVZzlq9BHnP4cYag~Iq2fkx9Tda31G2obbcEkw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Effect of baseline albuminuria on renal endpoint (combined endpoint of doubling of serum creatinine, ESRD, or death) and on the individual component of ESRD in the RENAAL study. Adjusted for baseline risk markers, the renal endpoint was 5-fold higher and risk of progression for ESRD 8-fold higher in the high versus the low albuminuria group [see 101].
Effect of baseline albuminuria on renal endpoint (combined endpoint of doubling of serum creatinine, ESRD, or death) and on the individual component of ESRD in the RENAAL study. Adjusted for baseline risk markers, the renal endpoint was 5-fold higher and risk of progression for ESRD 8-fold higher in the high versus the low albuminuria group [see 101].
Reducing Proteinuria
According to the second postulate, reducing proteinuria slows renal progression. While there is limited proof of concept from clinical interventional trials that specific titration against the level of proteinuria improves the efficacy of renoprotective therapy in most patients, many consider the ultimate goal of proteinuria remission (<1 g/day) to be valid [24]. Reducing proteinuria independent of other progression mechanisms would be essential to address the hypothesis that proteinuria causes progression of diabetic nephropathy [111]. Targeting proteinuria reduction in patients with established diabetic nephropathy in order to slow renal progression is generally accomplished with agents that reduce both proteinuria and blood pressure [112]. A broad range of therapeutic interventions to either reduce proteinuria or block the damaging effects of proteinuria (table 2) has been recently reviewed [111]. Data are very limited on therapies which might reduce proteinuria through other mechanisms unrelated to blood pressure reduction on RAS blockade [113, 114]. In the ACTION 1 study of the advanced glycation end product inhibitor pimagedine, significant reductions in proteinuria occurred compared to placebo, independent of changes in blood pressure, despite the nearly universal use of ACEI/ARBs in pimagedine- and placebo-treated patients. Patients with stable or decreased proteinuria experienced fewer instances of doubling of creatinine levels. However, the AGE inhibitor failed to produce a statistically significant overall reduction in the primary endpoint of doubling of serum creatinine in the study [113].

In nondiabetic kidney disease, data supporting a relationship between early reductions in proteinuria and slower renal progression have emerged, exclusively from secondary analyses of clinical trials [74, 115]. A recent post hoc analysis of the African American Study of Kidney Disease (AASK) trial has added new information [116] by assessing the initial reduction in proteinuria (6 months vs. baseline) as a predictor of decline in the glomerular filtration rate. Enrolled patients had been randomized to different levels of mean arterial pressure (102–107 vs. ≤92 mm Hg) and to different antihypertensive treatment groups. Similar to the relationship to the baseline level of proteinuria, the decline in glomerular filtration rate and risk of ESRD were strongly predicted by the initial change in proteinuria in this nondiabetic study of patients with hypertensive nephrosclerosis. The association of proteinuria change with subsequent progression of kidney disease was present regardless of baseline levels of proteinuria (an initial urine protein-to-creatinine ratio of >2.5 was an entry exclusion to the trial).
In the recently published COOPERATE-ABP substudy, about one third of patients enrolled in the original COOPERATE trial (randomized to ACEI, ARBs, or combination therapy) for nondiabetic kidney disease underwent ambulatory blood pressure monitoring [117]. Superior sustained proteinuria reduction (74%) in the combination group was associated with the least number of patients reaching the renal endpoints of creatinine doubling or ESRD.
Treatment of Hypertension
Hypertension affects up to 60% of diabetic patients and increases the risk of microvascular complications such as nephropathy [22, 112, 118]. Hypertension occurs early [119], and it is the major determinant of the rate of nephropathy progression [120]. Both cross-sectional and longitudinal studies have identified a correlation between hypertension and decline in renal function in type 2 patients [121,122,123], although the relationship between hypertension and type 1 and type 2 nephropathy may not be identical [124]. In patients with type 1 diabetes, the onset of microalbuminuria and its progression to overt nephropathy are associated with a higher level of blood pressure, and the relationship between hypertension and renal progression is clearly established [125]. In the majority of type 2 diabetic nephropathy patients, abnormal circadian blood pressure patterns or overt hypertension are already present prior to proteinuria. Compared to the normal renal vasoconstrictive autoregulatory response to hypertension, diabetic patients are less able to vasoconstrict the afferent arteriole, allowing hypertension to elevate glomerular capillary pressures, in association with greater degrees of proteinuria [125].
Early reports that effective antihypertensive treatment in diabetic nephropathy reduced albuminuria [126] and the risk of renal progression [127, 128] have been validated [129]. Aggressive early antihypertensive therapy reduces proteinuria in patients with type 1 diabetic nephropathy [21] and greater reductions may occur in patients who still have hyperfiltration [130]. With slowing of renal progression, proteinuria decreases, and the most significant reductions in proteinuria are associated with the largest reductions in blood pressure [131,132,133]. Following initiation of antihypertensive therapy in hypertensive patients with diabetic nephropathy, a faster initial reduction in glomerular filtration is followed by a slower sustained decline [134]. It should be noted that initiation of antihypertensive treatment results in an initial drop in function, regardless of the antihypertensive agent used. A recent ACEI study using varying doses of ramipril as the primary antihypertensive agent showed that proteinuria was reduced in a group with a mean arterial pressure of 92 mm Hg, but rose in the group with mean pressures only 8 mm Hg higher [135]. Reductions in systemic pressure are associated with lowering of glomerular capillary pressure. When systemic blood pressure is lowered 10–20%, ACE inhibitors, some calcium channel blockers (CCBs), and even conventional therapy with beta blockers or diuretics all have significant antiproteinuric effects [136, 137]. The most significant reductions in proteinuria have occurred in studies with the largest reductions in blood pressure [125]. However, the antiproteinuric effects of antihypertensive agents of different classes do vary considerably [21]. In contrast to significant reductions in protein excretion associated with ACEI/ARBs, for example, nondihydropyridine (NDHP) CCBs cause a milder decline, and dihydropyridine (DHP) calcium blockers and beta blockers no apparent reduction, apart from their hypotensive actions [138]. The different effects of classes of antihypertensive agents on proteinuria are likely due to different actions on hemodynamics or metabolism [136, 138]. Other modifying factors are the patient’s salt intake and levels of aldosterone in the circulation. While drug-specific effects may dominate pressure-dependent influences on proteinuria, patients responding to one class of drug frequently also benefit from other classes.
Angiotensin-Converting Enzyme Inhibitor
Beyond their antihypertensive benefit, ACEI slow the progression of type 1 diabetic nephropathy [139, 140]. The mechanisms of the beneficial nephroprotective activity of RAS inhibition in diabetic nephropathy are complex and still incompletely understood [24, 140, 141] and may vary somewhat between type 1 and type 2 diabetes. Operative actions (table 3) include systemic and intrarenal hemodynamic effects, improvements in the filtration barrier itself, and blockade of angiotensin II’s growth-stimulating actions that promote tissue fibrosis and extracellular expansion [142]. A recent human biopsy study indicated that renin-angiotensin components including the ACE and angiotensin II were present in tubulointerstitial compartments, indicative of elevated local angiotensin production associated with induction of proinflammatory parameters which were measured [143]. ACEI are known to reduce proteinuria in type 1 diabetic nephropathy by diminishing the size of large nonselective pores in the glomerular filtration barrier [144]. Recent data suggest that zona occludens-1, which plays a critical role in maintaining function of the epithelial slit diaphragm, is altered in experimental diabetic nephropathy, and that ACEI prevent the alteration [145]. In streptozotocin-treated rats with proteinuria, reduced nephrin gene expression is attenuated with ACE inhibition [146]. Other data indicate that ACEI do not correct the size-selective basement membrane dysfunction in type 2 diabetes, although other renal structural abnormalities, including interstitial expansion, may benefit [147].

Nearly 20 years have elapsed since the initial reports that converting enzyme inhibitors reduced severe proteinuria in diabetic nephropathy [148] independent of a fall in blood pressure. Subsequent studies have indicated that reduction in proteinuria is associated with slowing of renal progression in patients with overt nephropathy. ACEI reduce the level of proteinuria more than equivalent doses of other classes of antihypertensive agents do, similar to their impact on serum creatinine [149, 150] but the proteinuria advantage is lost as the systemic blood pressure declines [10, 136]. Event rates in clinical trial comparisons between ACEI and placebo become identical once treatments lower systemic blood pressure to means of less than 95 mm Hg [125]. The initial decrease in proteinuria with ACEI, which occurs within weeks of therapy, may be largely functional in nature. However, the nadir in proteinuria may require a year of ACEI therapy. A small subset of patients treated in a clinical trial setting appears to achieve proteinuria remission, and renal decline becomes nonprogressive [151].
The clinical benefit to reduce proteinuria appears to be less significant in type 2 diabetic nephropathy [42]. The results of ACEI remain inconclusive [104, 152, 153]. While beneficial effects on proteinuria have been demonstrated with ACEI, no prospective, controlled study has demonstrated a similar benefit on definite renal functional endpoints in type 2 patients [125]. Genetic variation in patients is one of many factors under evaluation to explain heterogeneous responses to RAS blockade [154]. Whether drug-related reductions or remissions in proteinuria can be interpreted as demonstrating ‘renal protection’ remains unclear, and a specific goal for proteinuria reduction has not been established by studies in either type 1 or type 2 diabetic nephropathy patients.
Angiotensin Receptor Blockers
Whether the ARBs were renoprotective in human diabetic nephropathy became the subject of two large clinical trials in the past decade. The differential effects of ARBs, in comparison to ACEI, have been reviewed elsewhere [155, 156]. Reduction of AT1 receptor stimulation by its ligand angiotensin II is the main action common to both ACEI and ARBs. The main differences are the blockade of bradykinin degradation by ACEI and the unopposed activation of the AT2 receptors when ARBs are utilized. Limited available data comparing the effects of ACEI and ARBs in diabetic nephropathy suggest similar effects on macroalbuminuria [157,158,159]. Comparative data on overt proteinuria are limited. Andersen et al. [158 ]reported comparable reduction of albuminuria between the ACEI enalapril and the ARB losartan in a double-blind, randomized crossover study in type 1 diabetic nephropathy. Additional investigation showed that the ARB reduced the abnormally elevated size-selective properties of the glomerular basement membrane in the diabetic kidney. AT1 receptor antagonists appear to be better tolerated than ACEI.
These differential effects of ARBs in comparison to ACEI formed the basis of the IDNT and RENAAL studies. In the RENAAL study, 1,513 patients with type 2 diabetic nephropathy were randomized to losartan or placebo for a mean of 3.4 years, in addition to conventional antihypertensive therapy [106]. The ARB reduced the incidence of serum creatinine doubling (risk reduction 25%) and ESRD (risk reduction 28%), and was associated with an average 35% reduction in proteinuria. Placebo-treated patients experienced a slight increase in the median change in proteinuria from baseline during the study. The initial (first 6 months) antiproteinura response to ARB therapy and the reduction in albuminuria due to losartan were recently reported [110]. Albumin was reduced by 28% from baseline in the losartan group in the first 6 months of the study, although the change in albumin in both groups was highly variable. Significant (23%) reduction in albuminuria was associated with a beneficial effect on renal protection even when correcting for other differences in risk factors. Furthermore, when residual albuminuria was adjusted for, the treatment effect of losartan on the renal endpoint was eliminated. These analyses of baseline and therapy-induced changes in albuminuria provide firm new evidence that albuminuria should be a primary target of therapy in type 2 diabetic patients with nephropathy.
The IDNT, the second randomized, double-blind, placebo-controlled study, also demonstrated the benefit of RAS inhibition with angiotensin II receptor blockers in hypertensive type 2 diabetic patients with overt nephropathy (900 mg proteinuria per day) [105]. The composite primary endpoint was doubling of the baseline serum creatinine level, development of ESRD (dialysis, transplantation, or serum creatinine ≥6.0 mg/dl), or death from any cause. Enrollment criteria were 1.0–3.0 mg/dl in females and 1.2–3.0 mg/dl in males. Study patients were taken off all ACEI, ARBs, and CCBs, and randomized to irbesartan 300 mg, amlodipine 10 mg daily, or placebo. After a mean follow-up of 2.6 years, irbesartan risk reduction was 20% versus placebo (p = 0.02) and 23% versus amlodipine (p = 0.006). Specific risk reduction for serum creatinine doubling was 33% with irbesartan versus placebo (p = 0.003) and 37% versus amlodipine (p < 0.001). Similar irbesartan risk reduction was found for ESRD progression, but not for mortality outcomes. Post hoc unpublished analysis indicated that risk reduction was related to baseline proteinuria: patients with nephrotic proteinuria had an event rate of 70% and a 14-fold increased risk of progression to advanced disease [154]. Mean baseline urinary protein excretion was 2.9 g/24 h in all groups. Treatment with irbesartan was associated with a 33% reduction in 24-hour urinary protein excretion, compared with 6% in the amlodipine group and 10% in the placebo group. Results of the RENAAL and IDNT studies demonstrating benefit of ARBs in progression of overt nephropathy in type 2 diabetes have led to regulatory drug approval for ARBs in initial therapy for hypertensive type 2 diabetic patients with proteinuric renal disease.
Combination Therapy
Effects to maximize the antiproteinuric effect of ACEI or ARBs include dietary salt restriction and addition of a diuretic [158]. The therapeutic advantage of combination ACEI/ARB therapy for nondiabetic renal disease has recently been reported [160]. The theoretical benefits of combination therapy include those of ACEI (decreased angiotensin II levels, increased bradykinin, depressed aldosterone) added to those of ARB therapy (blockade of angiotensin II produced by chymase, increased vasodilatory, antiproliferatative and antifibrotic effects) [161]. Examination of the RENAAL and IDNT data reveal that while renal progression was reduced, over 40% of those on losartan and over 30% of those on irbesartan reached primary endpoints [162]. With complementary biological mechanisms, dual blockade with ACEI and ARBs for diabetic proteinuria has theoretical advantages over monotherapy [163, 164].
In addition, in the nephropathy patient at high risk for cardiovascular complications, dual blockade offers cardiovascular as well as renal protection. Both ACEI and ARBs are associated with improved cardiovascular outcomes in clinical trials. Several studies, summarized recently [165], analyzing the effects of RAS inhibition on the risk of cardiovascular complications in study populations that included diabetics, have been reported. Favorable results on a composite of major cardiovascular events has been shown with ACEI [166]while similar protection may be achieved with ARB therapy [167,168,169]. Limited data are available comparing cardiovascular benefits of ACEI versus ARBs [170, 171]. Until the issue is resolved, a strategy of dual blockade has been suggested [165].
Little is known about the comparable effects of ACEI and ARBs on proteinuria in patients with overt diabetic nephropathy [172, 173]. Data in nondiabetic renal disease [174] suggest that combination therapy might synergistically reduce proteinuria levels. However, data comparing dual blockade with equivalent blocking doses of either class, or with the ARB/ACEI used with another agent, in groups with comparable blood pressures, are not available. It is likely that combination therapy will preclude use of maximal doses of either class of agents in the clinical setting [175].
Calcium Channel Blockers
While ACEI and ARBs are the most effective antiproteinuria agents for diabetic nephropathy, additional therapy may be necessary to meet the currently recommended blood pressure targets. In addition, a rise in serum creatinine of more than 35% above baseline or the emergence of hyperkalemia may be a reason to withhold first-line ACEI or ARB therapy [176, 177]. CCBs are among the most commonly used classes of antihypertensive drugs in patients with kidney disease [178]. However, evidence for any renoprotective effects of calcium antagonists is equivocal. DHP and NDHP CCBs have differential effects on proteinuria. A systematic review of 28 randomized clinical trials in hypertensive proteinuric adults with or without diabetes that included a DHP or NDHP treatment arm was recently published by Bakris et al. [179]. NDHP CCBs alone or in combination with an ACEI or ARB produced significant reductions in proteinuria, compared to no antiproteinuric effects in DHP CCBs, in the presence or absence of diabetes. NDHP CCBs appear to have properties that affect glomerular permselectivity differently than DHP CCBs [180], not directly related to blood pressure reductions. When used in combination with ACEI in type 2 diabetes, NDHP CCBs induce a greater reduction in proteinuria than DHP CCBs [181]. A recent study in nondiabetic nephropathy of either the NDHP verapamil or the DHP amlodipine with an ACEI suggested that the DHP CCB had an adverse effect on selectivity of proteinuria [178].
The Surrogate Endpoint
According to the third postulate of the proteinuria hypothesis, proteinuria is a potential surrogate endpoint for current outcome measures [182, 183], and diabetic nephropathy is a disease model for the potential use of proteinuria as a surrogate [2]. In chronic kidney disease, trials to evaluate new drug treatments have traditionally been required to use recognized ‘hard’ endpoints of dialysis, transplantation, and death rates, for regulatory approval [184]. These have been combined more recently with the ‘first generation’ surrogate marker, doubling of serum creatinine, over the course of the trial. The use of the serum creatinine surrogate has allowed studies to show benefit with smaller sample sizes over a reduced drug exposure time [185] even at an intermediate stage of disease severity, where composite ‘hard’ endpoints less frequently occur. However, because changes in serum creatinine are less sensitive in the earliest stages of kidney disease progression, prohibitively large early intervention trials of longer duration will again be required [186], making creatinine impractical as the sole outcome marker.
Insofar as early intervention is critical [26] in diabetic nephropathy, a valid earlier (‘second generation’) surrogate marker with effects proportional to accepted endpoints would be particularly valuable [12]. While it is an appropriate measure in patients with more advanced disease undergoing decline in renal function over months, the creatinine rise may be prohibitively slow in early stages of diabetic nephropathy. Furthermore, disease progression as measured by creatinine doubling may be slowest, with low event rates, in the earliest stages of the disease, when intervention might be most successful. The selection of a surrogate outcome would require a clinical connection between the surrogate and the established outcome in the context of the disease and intervention under investigation.
In the case of proteinuria, disadvantages include the intrasubject variability in proteinuria, uncertainty regarding meaningful reduction in proteinuria, and the lack of ‘breakthrough’ drugs with isolated antialbuminuric or antiproteinuric effects to be tested. Studies utilizing proteinuria as a primary outcome measure linked to secondary harder endpoints for new drug approval are currently lacking. The above analysis indicates that the relationship of albuminuria and proteinuria to the course of diabetic nephropathy is complex. Strict interpretation of available data on RAS blockade does suggest that albumin should be reduced to minimal achievable levels in diabetic nephropathy. While proteinuria reduction would be advantageous as a new surrogate outcome measure to facilitate regulatory drug evaluation, this postulate is likely to depend on factors specific to the diseases and the drugs used.
Conclusion
Overt diabetic nephropathy is characterized by proteinuria and progressive loss of kidney function, and treatment to delay progression now includes strict control of proteinuria. Complex mechanisms altering the protein-selective glomerular basement membrane exist in diabetic nephropathy, and form the targets of current therapies. Large clinical studies support an overall relationship between severity of albuminuria or proteinuria and rate of progression in diabetic nephropathy. The benefit of albuminuria reduction, at least by RAS blockade, to minimum goals in diabetic nephropathy, is emerging. While diabetic kidney disease is a paradigm for proteinuria as a surrogate marker for renal function in the drug approval process, this shift in perspective will require prospective data from additional clinical trials, particularly of non-RAS drugs, to be complete. More needs to be learned about human diabetic nephropathy before the proteinuria hypothesis can be fully adopted.
Acknowledgments
The author wishes to express gratitude to Dr. Richard Solomon and Dr. Franklin H. Epstein for their expert review of the manuscript.
