

Abstract
Longitudinal bone growth occurs at the growth plate by endochondral ossification. Within the growth plate, chondrocyte proliferation, hypertrophy, and cartilage matrix secretion result in chondrogenesis. The newly formed cartilage is invaded by blood vessels and bone cells that remodel the newly formed cartilage into bone tissue. This process of longitudinal bone growth is governed by a complex network of endocrine signals, including growth hormone, insulin-like growth factor I, glucocorticoid, thyroid hormone, estrogen, androgen, vitamin D, and leptin. Many of these signals regulate growth plate function, both by acting locally on growth plate chondrocytes and also indirectly by modulating other endocrine signals in the network. Some of the local effects of hormones are mediated by changes in paracrine factors that control chondrocyte proliferation and differentiation. Many human skeletal growth disorders are caused by abnormalities in the endocrine regulation of the growth plate. This review provides an overview of the endocrine signals that regulate longitudinal bone growth, their interactions, and the mechanisms by which they affect growth plate chondrogenesis.
Introduction
Longitudinal bone growth occurs at the growth plate by a process called endochondral ossification in which cartilage is first formed and then remodeled into bone tissue. The growth plate consists of three principal layers: the resting zone, proliferative zone, and hypertrophic zone (fig. 1). Chondrocyte proliferation, hypertrophy, and extracellular matrix secretion result in chondrogenesis. The newly formed cartilage is invaded by blood vessels and bone cell precursors, which remodel the hypertrophic zone cartilage into bone. The net effect is that new bone tissue is progressively created at the bottom of the growth plate, resulting in bone elongation.

Histology of the growth plate. The growth plate is a thin cartilage structure situated in the ends of tubular bones. It is commonly subdivided into three distinct zones; the resting, proliferative, and hypertrophic zones.
Histology of the growth plate. The growth plate is a thin cartilage structure situated in the ends of tubular bones. It is commonly subdivided into three distinct zones; the resting, proliferative, and hypertrophic zones.
The growth plate contains one cell type, the chondrocyte, at different stages of differentiation. Resting zone chondrocytes replicate at a slow rate [1] and act as the stem-like cells that replenish the pool of proliferative chondrocytes [2]. Proliferative zone chondrocytes replicate at a high rate [1], and the resulting daughter cells line up along the long axis of the bone. As a result, clones of chondrocytes are arranged in columns parallel to this axis, a process critical to the formation of bones with an elongated shape [2].
At a certain point, the cells stop dividing and terminally differentiate into hypertrophic chondrocytes [1]. During the hypertrophic process, chondrocytes increase their height about 6- to 10-fold, and thus hypertrophic differentiation makes an important contribution to longitudinal growth [3]. The hypertrophic chondrocytes calcify the surrounding extracellular matrix and produce factors that attract the invading bone cells and blood vessels, including vascular endothelial growth factor [4]. Hypertrophic chondrocytes undergo apoptosis shortly before the blood vessels invade the chondrocyte lacuna [5].
Basic Principles of Growth Plate Physiology
The rate of growth plate chondrocyte proliferation, and hence the rate of longitudinal bone growth, falls progressively with age [1]. That decrease in growth plate function appears to be due to a mechanism intrinsic to the growth plate rather than a hormonal or other systemic mechanism; in growth plate transplantation experiments, the growth rate of the transplanted growth plate depends on the age of the donor animal, not the age of the recipient [6]. The term growth plate senescence [7] has been used to describe this intrinsic process that involves both a decline in the function and cellularity of the growth plate. Recent evidence suggests that this decline occurs because stem-like cells in the resting zone have a finite proliferative capacity that is gradually exhausted [8, 9]. In some mammals, including humans, the growth plate cartilage is completely replaced by bone at the time of sexual maturation. This event, termed epiphyseal fusion, appears to be triggered when the proliferative capacity of the growth plate chondrocytes is finally exhausted [8, 9].
Longitudinal bone growth is tightly governed by complex endocrine controls such that optimal growth occurs only in a healthy, well-nourished individual. If the individual is ill or malnourished, insulin-like growth factor (IGF)-I levels decline, glucocorticoid levels may rise, and thyroid hormone levels may decline causing linear growth to slow. Thus, one of the teleological reasons for the endocrine regulation may be to conserve nutrients for vital functions during times of adversity. At such times growth may be an unaffordable luxury that must be postponed until better times. Indeed, when this occurs, growth is primarily delayed and not lost irreversibly.
Our understanding in this field is derived primarily from two sources. First, human diseases involving hormone deficiency, hormone resistance, and hormone excess have provided great insight into the physiological role of hormones in human skeletal growth. Second, animal studies, including gland ablation, hormone administration, and molecular ablation or overexpression of specific endocrine-related genes, have also provided insights. However, findings in other mammals may not apply directly to humans. For example, the stimulatory effects of estrogen on skeletal growth and maturation, which occur in humans, are poorly reproduced in rodent studies.
Growth Hormone and Insulin-Like Growth Factors
Growth hormone (GH) and insulin-like growth factors (IGFs) are potent stimulators of longitudinal bone growth. GH excess, due to pituitary adenomas in childhood, results in gigantism. Conversely, GH deficiency or insensitivity due to GH-receptor mutations or defects in GH-signaling pathways markedly impairs postnatal growth [10,11,12]. Final height data on patients with untreated isolated GH deficiency suggests that it leads to an average final height SDS of –4.7 (–6.1 to –3.9) [12]. GH deficiency and insensitivity do not impair prenatal growth significantly. In contrast, both pre- and postnatal growth deficits occurred in the only reported cases of IGF-I deficiency due to a mutation in the igf-1 gene [13] and of IGF-I resistance due to mutations in the type I IGF receptor gene [14].
The role of the GH-IGF axis in longitudinal bone growth has been evaluated by genetic targeting of its components in mice. Mice lacking the GH gene exhibit normal birth weight, but a reduction in postnatal growth [15]. Mice lacking either the igf-1 or igf-2 genes show intrauterine growth retardation with birth weights approximately 60% that of wild-type littermates [16]. Mice deficient in the IGF-I receptor have even more severe reduction in birth weight (45% of wild type) and die soon after birth due to respiratory failure [16].
The original somatomedin hypothesis stipulates that the effect of GH on linear growth is mediated by liver-derived IGF-I [17]. The role of circulating IGF-I has been evaluated recently using tissue-specific gene targeting. Liver-specific ablation of igf-1 in mice using the cre-lox system reduces circulating IGF-I levels by approximately 80%, but has no appreciable effect on postnatal growth. However, in these mice, changes in IGF-binding proteins may have preserved bioactive IGF-I levels [18]. Combined deficiency of liver-derived IGF-I and knock-out of acid-labile substance (a component of the circulating IGF-I complex) further reduces circulating IGF-I levels and does inhibit linear growth and growth plate height [19]. In addition, in both mice and humans with inactivating mutations in the GH receptor, increased circulating IGF-I can markedly improve linear bone growth, supporting a role for circulating IGF-I [20, 21].
GH can also stimulate longitudinal bone growth by a local action on the growth plate. Injection of GH into the tibial growth plate accelerates longitudinal growth in the injected growth plate compared to the vehicle-injected contralateral growth plate [22]. Part of the local action of GH on the growth plate may be mediated by increased local production of IGF-I, which then acts in a paracrine/autocrine fashion to increase chondrogenesis [23, 24]. In addition, GH may also have an effect on the growth plate that is independent of both endocrine and paracrine IGF-I. The dual effector hypothesis states that GH acts locally at the growth plate to recruit resting chondrocytes into a proliferative state [25,] as well as to stimulate local IGF-I production, which then stimulates proliferation of proliferative zone chondrocytes [24,25,26]. However, detailed in vivo labeling experiments suggest that IGF-I, like GH, can stimulate proliferation of resting zone chondrocytes and chondrocyte hypertrophy [27]. In contrast, studies in IGF-I-deficient mice suggest that IGF-I acts primarily to increase hypertrophic cell height, with little effect on proliferation [28].
Glucocorticoid
Glucocorticoids are widely used as anti-inflammatory and immunosuppressive drugs in children. Long-term, high-dose glucocorticoid treatment often leads to growth failure. Similarly, systemic administration of glucocorticoid in mice, rats, and rabbits decreases the rate of longitudinal bone growth, by inhibiting growth plate chondrocyte proliferation [8, 29]. In addition, glucocorticoid may stimulate apoptosis of growth plate chondrocytes [29, 30].
Glucocorticoid inhibits longitudinal bone growth, in part, through a direct effect on growth plate chondrocytes. Glucocorticoid receptor is expressed by growth plate chondrocytes [29]. Furthermore, glucocorticoid inhibits proliferation of growth plate chondrocytes in vitro [31]. In addition, local infusion of dexamethasone into the growth plate causes a local inhibition of longitudinal bone growth [32].
The local effects of glucocorticoid on growth plate chondrocyte proliferation may be mediated, in part, by changes in the local IGF-I system. Short-term systemic administration of glucocorticoid in rodents decreases IGF-I expression in the growth plate [33]. In contrast, long-term treatment of rodents increases IGF-I expression in the growth plate [34] and growth-suppressive doses of dexamethasone given to rabbits actually increases GH-receptor mRNA expression in the growth plate [35]. In vitro, glucocorticoid suppresses GH-receptor expression in cultured rat growth plate chondrocytes, while type I IGF receptor expression is not affected [31].
In addition to its direct action on the growth plate, glucocorticoid may also suppress longitudinal bone growth through an indirect action, involving other endocrine signals. In humans, glucocorticoid excess has been associated with reduced GH secretion in some studies [36], but not all [37]. The interpretation of these findings is complicated by the presence of the underlying disease requiring glucocorticoid treatment and by the suppressive effect of obesity per se on GH secretion. In humans, plasma IGF-I and IGF binding protein 3 levels have been found to be either normal [36] or increased [38].
Discontinuation of glucocorticoid treatment is followed by catch-up growth [8], which is caused, at least in part, by a mechanism intrinsic to the growth plate [7]. Catch-up growth may occur because the decreased cell proliferation during glucocorticoid treatment conserves the proliferative capacity of the chondrocytes, thus slowing growth plate senescence. Following discontinuation of the glucocorticoid treatment, the growth plates are less senescent than normal, and hence show a greater growth rate and grow for a longer time than expected for age, resulting in catch-up growth [8]. Despite catch-up growth, prolonged glucocorticoid administration in children can result in some residual permanent growth deficit.
Thyroid Hormone
Thyroid hormone is necessary for normal skeletal growth and maturation. Hypothyroidism slows longitudinal bone growth and endochondral ossification, while hyperthyroidism accelerates both processes [39, 40]. In hypothyroid animals, there is a decrease in the heights of the proliferative and hypertrophic zones, and a decrease in chondrocyte proliferation, chondrocyte hypertrophy, and vascular/bone cell invasion [41]. In addition, the normal columnar organization of the growth plate is disrupted [41].
Some of the skeletal effects of thyroid hormone appear to be due to a direct action on the growth plate. In fetal mouse tibia organ culture, thyroid hormone promotes longitudinal growth with the largest effect seen in the hypertrophic zone [42]. In cell culture, thyroid hormone stimulates hypertrophic differentiation, but often diminishes proliferation [43]. Local conversion of T4 to T3 by thyroid hormone deiodinase 2 in the growth plate may contribute to local effects [42].
Growth plate chondrocytes express thyroid hormone receptor (TR) isoforms TR-α1, α2, and β1[44]. Knocking out TR-β isoforms in mice has little effect on the skeleton. In contrast, ablation of TR-α impairs longitudinal bone growth and endochondral ossification, effects that resemble hypothyroidism [45]. In humans, one family with homozygous deletion of TR-β showed epiphyseal stippling and some delayed skeletal maturation but normal growth, suggesting that TR-β mediate some of the effects of thyroid hormone on human skeletal development [46]. Thus, deletion of TR-β affects skeletal development in humans, but has little effect in mice. Most cases of thyroid hormone resistance in humans are caused by dominant-negative mutations of the TR-β gene that may affect TR-α function as well, and show variable skeletal effects [46].
In addition to its local action on the growth plate, thyroid hormone may have indirect effects on the growth plate, mediated by GH and IGF-I. In hypothyroid humans and mice, GH and IGF-I levels are reduced [47]. Replacing GH in hypothyroid rats, or in mice lacking TR-α, improves longitudinal bone growth. However, GH does not normalize growth plate endochondral ossification or morphology [47].
Estrogen
Premature estrogen exposure, as in precocious puberty, accelerates skeletal maturation, thus causing premature epiphyseal fusion and decreased final height. Conversely, lack of estrogen, as in hypogonadism, results in delayed fusion and tall stature. The role of estrogen in skeletal maturation and epiphyseal fusion was verified by the discovery of an estrogen-resistant man with a homozygous null mutation in the estrogen receptor-α (ER-α) gene [48], and the detection of adults and children of both sexes with estrogen deficiency, as a consequence of mutations in the aromatase gene [49]. These patients exhibited normal growth until puberty, but failed to undergo epiphyseal fusion during puberty. Decelerating linear growth persisted into adulthood, resulting in tall stature. Thus, estrogen signaling through ER-α is crucial for normal skeletal maturation and epiphyseal fusion. These findings suggest that the final height of children might be increased by therapy with aromatase inhibitors [50] or antiestrogens [51]. Selective ER modulators could serve either as estrogen agonists or antagonists at the growth plate [52, 53].
Indirect evidence suggests that epiphyseal fusion occurs when the proliferative capacity of the growth plate chondrocytes is exhausted [9] and that estrogen acts by advancing growth plate senescence, causing earlier proliferative exhaustion, and thus earlier fusion [9]. This concept would explain why estrogen exposure does not induce fusion rapidly, but often must act for years before fusion occurs, particularly in young children.
During puberty, sex steroids induce a pubertal growth spurt. This growth acceleration may be primarily induced by estrogen since a near-normal growth spurt occurs in patients with androgen insensitivity [54], whereas little or no growth spurt appears to occur in patients with aromatase deficiency [55]. In addition, low-dose estrogen treatment can accelerate growth in both prepubertal boys and girls [56], and the accelerated growth in patients with familial male-limited precocious puberty is curtailed by the administration of an aromatase inhibitor [57]. The pubertal growth spurt also has a better temporal correlation with the increase in estrogen levels than with the increase in androgen levels [58]. Much of the growth acceleration appears to be mediated by estrogen-induced stimulation of the GH-IGF-I axis [55].
In addition to its effect on the GH-IGF-growth plate axis, estrogen also may act directly on the growth plate. Three lines of evidence support this hypothesis. First, ER-α and -β are expressed in growth plates cartilage [59]. Second, estrogen inhibits longitudinal bone growth of hypophysectomized and castrated female rats [60]. Third, human growth plate chondrocytes placed in primary culture reportedly respond to estrogen stimulation [61].
Gunther et al. [51] were able to inhibit estrogen-induced bone maturation using ICI 182,780, a pure antiestrogen, thus suggesting that estrogen acts to advance bone maturation, at least in part, through a classical ER.
The effects of estrogen on the growth plate differ markedly in mice and humans. First, mice, like most mammals studied, lack a pubertal growth spurt. In mice, estrogen does not stimulate longitudinal bone growth, but rather inhibits growth, an effect seen only at high doses in humans. Second, mice do not undergo epiphyseal fusion at the time of sexual maturation. Estrogen may promote fusion in mice, but only at supraphysiological concentrations. Third, in mice, the effects of estrogen on the growth plate appear to be mediated primarily by ER-β rather than ER-α. Female mice lacking ER-β have slightly longer femurs than wild-type females [62]. In contrast, female mice lacking ER-α show decreased longitudinal bone growth and earlier epiphyseal fusion, perhaps because the increased levels of circulating estrogen in these mice act through ER-β to accelerate epiphyseal fusion. Female mice lacking both ER-α and ER-β have slightly elevated circulating estrogen, but do not exhibit accelerated epiphyseal fusion [63].
Androgen
Androgen also contributes to the pubertal growth spurt. Some of this effect is probably due to aromatization of androgens to estrogens in various peripheral tissues, including adipose tissue. Aromatase, the enzyme that converts androgens to estrogens, is expressed in growth plate cartilage [64], suggesting that some of the effects of androgen may be due to local conversion into estrogen. However, androgen per se, without conversion to estrogen, also appears to stimulate longitudinal bone growth. In boys, dihydrotestosterone, a nonaromatizable androgen, can accelerate linear growth [65]. This effect is not associated with increased circulating GH or IGF-I [65]. Similarly, testosterone stimulates growth in the absence of GH in hypophysectomized and castrated rats [60, 66].
These GH-independent effects of androgen may be due to a direct action of androgen on growth plate chondrocytes. Androgen receptor expression has been detected in rat [67] and human growth plate cartilage [59, 68]. Local administration of testosterone reportedly increases unilateral rat tibial epiphyseal growth plate width [69]. Furthermore, in vitro, dihydrotestosterone can stimulate proliferation and proteoglycan synthesis in growth plate chondrocytes [70]. Similarly, testosterone, and to a lesser extent, dihydrotestosterone stimulates chondrocyte proliferation in the mouse mandibular condyle, an organ culture model of endochondral ossification [71]. These local effects may be mediated, in part, by increased local IGF-I expression [70, 71].
Vitamin D
In growing mammals, vitamin D deficiency produces a constellation of skeletal abnormalities known as rickets. The width of the hypertrophic zone of the growth plate is increased and mineralization is defective [72]. Similar effects occur in mice lacking the vitamin D receptor. In these mice there is decreased apoptosis of hypertrophic chondrocytes at the metaphyseal ends of the columns and delayed invasion by blood vessels and bone cells [73]. Rickets also occurs in hypophosphatemic mice and humans, suggesting that the decreased extracellular phosphate, which occurs in vitamin D deficiency, may play a key role in rickets. Indeed, a mineral-enriched diet can normalize the growth plates of vitamin D-deficient and vitamin D-resistant animals [74, 75]. Thus the growth plate effects of vitamin D may be mediated primarily through the vitamin D receptor expressed in intestinal epithelial cells, leading to increased calcium and phosphate absorption from the intestinal lumen.
Vitamin D metabolites, however, may also act directly on the growth plate. 24,25-dihydroxyvitamin D3 injected directly into rachitic chick growth plates resulted in healing [76]. In vitro, 24,25-dihydroxyvitamin D3 stimulates differentiation but decreases proliferation in resting zone cells, while 1,25-dihydroxyvitamin D3 decreases proliferation in resting and proliferative zones [77]. These effects appear to be mediated by a signal transduction pathway in chondrocytes involving cell surface receptors, phospholipases, prostaglandins, and the mitogen-activated protein kinase cascade [78]. Local conversion of vitamin D metabolites also occurs in rat costochondral chondrocytes by 1α-hydroxylase and 24-hydroxylase [79]. The physiological role of local vitamin D action on the growth plate remains unclear.
Leptin
Leptin, a protein secreted primarily by white adipose tissue, regulates food intake and body weight. Because circulating leptin levels are increased in obesity, it has been hypothesized that leptin might contribute to the robust linear growth that occurs in obese children, despite low GH levels after craniopharyngioma surgery, and in idiopathic obesity [80]. Consistent with this hypothesis, leptin deficiency in mice impairs longitudinal bone growth, while treatment of these mice with leptin injections increased bone growth [81]. In contrast, in the few humans described with leptin deficiency or leptin-receptor deficiency, skeletal growth appeared grossly normal [82, 83]. With in vivo studies, it is difficult to distinguish between leptin’s direct effect on bone from its other systemic effects on nutritional balance, the GH-IGF axis, and circulating glucocorticoid levels. In organ cultures of mandibular condyle, a model of endochondral ossification, chondrocytes were found to contain specific binding sites for leptin [84], and leptin, at high concentrations, was found to stimulate chondrocyte proliferation and differentiation as well as IGF-I-receptor expression. Passive immunization of IGF-I abolished the effects of leptin on cell proliferation and differentiation [84]. The presence of the leptin receptor was also reported in primary cell culture of rabbit chondrocytes [85]. Taken together, these lines of evidence suggest that leptin may have a direct effect on chondrocytes in addition to its indirect effects, exerted through its ability to regulate appetite, energy metabolism, and endocrine systems. Leptin appears to affect bone density in mice by a mechanism involving the sympathetic nervous system [86]. Whether leptin can regulate longitudinal bone growth by a similar mechanism is not known.
Conclusions
Longitudinal bone growth at the growth plate is governed by a complex network of endocrine signals. Most of these signals regulate growth plate function by acting locally on growth plate chondrocytes and also indirectly by modulating other endocrine signals in the network (fig. 2). Some of the local effects of hormones are mediated by changes in paracrine factors that control chondrocyte proliferation and differentiation.
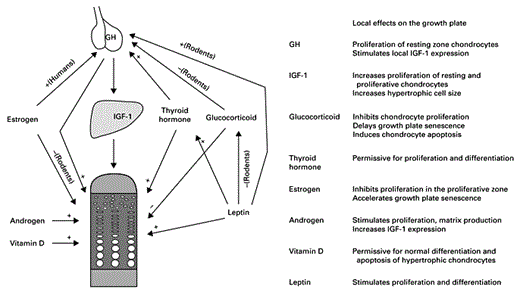
Endocrine signals that regulates longitudinal bone growth. Arrows indicate direct action on the growth plate and indirect action by modulating other endocrine signals. + = Stimulatory effect; – = inhibitory effect.
Endocrine signals that regulates longitudinal bone growth. Arrows indicate direct action on the growth plate and indirect action by modulating other endocrine signals. + = Stimulatory effect; – = inhibitory effect.
Our improved understanding in this field has provided insight into the molecular genetic causes of human growth failure. For example, human mutations affecting the GH-IGF-I system at many different levels have recently been shown to cause short stature, including mutations in genes encoding the GH-releasing hormone receptor [87], GH [88], STAT5b [10], IGF-I [13], acid labile subunit [89], and IGF-I receptor [14]. Molecular genetic defects affecting other endocrine regulators of growth also can cause growth plate dysfunction and short stature but often in conjunction with dysfunction of other tissues and organs, for example, mutations involving TR-β [46].
New experimental approaches, including tissue-specific gene targeting, microarray analysis, and linkage studies exploiting the full sequence of the human and mouse genomes promise to accelerate progress in understanding these regulatory systems. These advances will lead to improved understanding of human skeletal growth disorders and are likely to yield new therapeutic approaches.
