

Abstract
Hypogonadotrophic hypogonadism (HH) is characterized by delayed or absent pubertal development secondary to gonadotrophin deficiency. HH can result from mutations of the gonadotrophin-releasing hormone receptor 1, the gonadotrophin β-subunits, or various transcription factors involved in pituitary gland development. HH occurs in DAX1 mutations when associated with adrenal insufficiency (adrenal hypoplasia congenita), and is also linked with obesity in patients with mutations of leptin and its receptor, as well as mutations in prohormone convertase 1. Rarely, HH has resulted from kisspeptin receptor (GPR54) mutations, a gene implicated in the regulation of pubertal onset. When occurring with anosmia (a lack of sense of smell), HH is referred to as Kallmann’s syndrome (KS). Two KS-related loci are currently known: KAL1, encoding anosmin-1, responsible for X-linked KS, and KAL2, encoding the fibroblast growth factor receptor 1 (FGFR1), mutated in autosomal dominant KS. Anosmin-1 is an extracellular glycoprotein with some unique structural characteristics; it interacts with both urokinase-type plasminogen activator and FGFR1. It has previously been shown that anosmin-1 enhances FGFR1 signalling in a heparan sulphate-dependent manner, and proposed that anosmin-1 fine-tunes FGFR1 signalling during olfactory and GnRH neuronal development. Here, we review the known normosmic causes of HH, and discuss novel developmental and molecular mechanisms underlying KS; finally, we introduce three novel genes (NELF, PKR2, and CHD7) that may be associated with some phenotypic features of KS.
Introduction
In humans, pulsatile release of hypothalamic gonadotrophin-releasing hormone (GnRH) is essential for functioning of the hypothalamic-pituitary-gonadal (HPG) axis. GnRH is secreted into the hypophyseal portal circulation of the median eminence, where it activates the GnRH receptor 1 (GnRHR1) on the anterior pituitary gonadotrophs, thereby stimulating expression, synthesis and secretion of the two pituitary gonadotrophins: luteinizing hormone (LH) and follicle-stimulating hormone (FSH). LH and FSH stimulate steroid production and gametogenesis in both sexes. Production of gonadotrophins and resulting reproductive competence are therefore dependent on correct development and coordinate functioning of both hypothalamic GnRH-secreting cells and pituitary gonadotrophs [1, 2].
During human embryogenesis, GnRH-secreting neurons migrate from the olfactory placode to the mediobasal hypothalamus, where they undergo terminal differentiation and GnRH secretion [3, 4]. These migratory steps are strictly regulated by specific spatiotemporal expression patterns of axonal guidance cues, cell adhesion molecules and extracellular matrix (ECM) proteins, as well as specific transcription and growth factors and neurotransmitters that determine GnRH neuronal fate [5]. GnRH secretion is temporarily activated postnatally (for 3–6 months in the male), but then remains quiescent until the onset of puberty when the HPG axis is reawakened and secondary sexual maturation begins.
Hypogonadotrophic hypogonadism (HH) is a disorder of the HPG axis, characterized by failure of gonadotrophin secretion presenting as complete or partial failure of pubertal development. Clinical presentation also includes micropenis and cryptorchidism in the male, and amenorrhoea in the female. HH may be either congenital or acquired, secondary to hypothalamic or pituitary lesions. Congenital HH may be isolated, or occur in association with craniofacial anomalies/midline defects, and in certain disorders such as the Prader-Willi syndrome. Although most cases of HH are ‘idiopathic’, the last few decades have witnessed the gradual unravelling of the genetic basis of an increasing number of specific aetiologies. These advances have played a significant role in elucidating the pathophysiology of HH, and also in unfolding the complexities of the HPG axis.
Congenital HH is often classified into two categories: one, associated with anosmia (a lack of sense of smell: Kallmann’s syndrome (KS)), the other, associated with normosmia. Additional neurological and non-neurological signs may co-exist, depending on the specific mode of inheritance. The application of conventional linkage studies to investigate the genetic basis of HH has proven difficult, both because of its rarity and the infrequency of familial transmission; more importantly, pedigrees tend to be small since the majority of patients, without treatment, remain infertile [6]. Three modes of inherited KS are described: X-linked (KAL1, OMIM:308700), autosomal dominant (KAL2, OMIM:147950), or autosomal recessive (KAL3, OMIM:244200), with only KAL1 and KAL2 hitherto cloned. We first provide a molecular genetic overview of normosmic forms of HH, including those caused by mutations of GnRHR1, the gonadotrophin β-subunits (LH β and FSH β), pituitary transcription factors (HESX1, LHX3, and PROP1), DAX1, leptin and its receptor (LEP and LEPR), prohormone convertase 1 (PC1), GPR54, and KiSS1, and then discuss recent insights into the pathogenesis of KS (fig. 1).
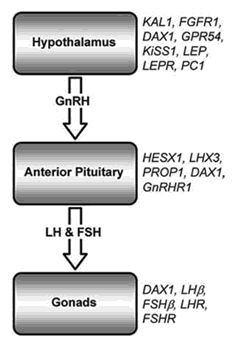
Schematic diagram of the HPG axis. The genes which are mutated in some patients with heritable forms of hypogonadism are indicated on the right. Mutations in all of these genes (except LHR and FSHR) are responsible for some cases of HH, and are discussed in more detail in this review, with particular emphasis on the KS genes (KAL1 and FGFR1). LHR = LH receptor; FSHR = FSH receptor.
Schematic diagram of the HPG axis. The genes which are mutated in some patients with heritable forms of hypogonadism are indicated on the right. Mutations in all of these genes (except LHR and FSHR) are responsible for some cases of HH, and are discussed in more detail in this review, with particular emphasis on the KS genes (KAL1 and FGFR1). LHR = LH receptor; FSHR = FSH receptor.
Normosmic HH
GnRH and Its Receptor
Although an obvious candidate, no GnRH mutations causing HH have been reported, in contrast to a recessive HH mouse model due to a large intragenic GnRH deletion [7] that has previously been defined. GnRHR1 mutations are seen in 40% of autosomal recessive normosmic HH patients [8, 9], the heterozygote being in general unaffected. GnRHR1 mutations often lead to impaired GnRH binding and/or receptor activation, with a consequential reduction in pituitary GnRH responsiveness. There is a broad spectrum of phenotypes in patients with GnRHR1 mutations, even within the same kindred, ranging from complete to partial HH [reviewed in [2]], suggesting the involvement of modifier genes, epigenetic factors, or environmental mechanisms.
Gonadotrophins (LH and FSH)
Gonadotrophins are heterodimers of a specific β-subunit with an α-subunit common to all glycoprotein hormones (TSH, LH, FSH, and hCG). No human α-subunit mutations have been reported, probably because such mutations would be non-viable [1]. However, mutations in the FSH and LH β-subunit genes have been reported in patients with delayed puberty [10, 11].
Pituitary Transcription Factors
The development of the normal adenohypophysis results from the precise spatial and temporal interaction of several signalling molecules including fibroblast growth factor 8, sonic hedgehog, and bone morphogenetic proteins 2 and 4, acting in concert with the homeobox transcription factors LHX3, PROP1, and HESX1 [12]. Mutations in LHX3, PROP1, and HESX1 cause some cases of combined pituitary hormone deficiency (CPHD), which are often associated with a number of midline abnormalities [2]. Netchine et al. [13] reported a homozygous LHX3 mutation in two unrelated consanguineous families, who presented with CPHD (but normal corticotroph function) that notably included pituitary deficits in LH and FSH. Three of the oldest patients failed to show any signs of spontaneous puberty by the age of 15 years. CPHD patients with PROP1 mutations demonstrate variable degrees of gonadotrophin insufficiency, from mild (occasionally late onset) to extremely severe, in association with lactotroph, somatotroph, and thyrotroph deficiency at birth, and occasional progressive corticotroph deficiency [14, 15]. Dattani et al. [16] showed that some cases of septo-optic dysplasia (SOD) were associated with homozygous mutations of HESX1. SOD is a condition defined by any combination of optic nerve hypoplasia, pituitary gland hypoplasia and midline abnormalities of the brain such as absence of the corpus callosum and septum pellucidum. SOD varies greatly in its severity and may include either sexual precocity or pubertal failure. Heterozygous mutations of HESX1 have been associated with SOD with milder CPHD [17].
HH with Adrenal Hypoplasia Congenita
HH occurs in conjunction with adrenal hypoplasia congenita in patients with loss-of-function mutations in the DAX1 (NR0B1) gene, encoding the orphan nuclear receptor, DAX1 [18, 19]. Steroidogenic factor-1 (SF1) is a further orphan nuclear receptor that regulates the transcription of genes involved in the adrenal and gonadotrophic axes. Sf1 knockout mice show complete adrenal and gonadal agenesis [20], and some patients with mutations in the gene encoding SF1 have XY sex reversal with adrenal failure [21]. DAX1 antagonizes SF1-mediated transcription in vitro, but they have both been shown to act independently or cooperatively during male gonadal development [22].
HH with Obesity
Mutations both of the leptin gene (LEP) and its receptor (LEPR) are associated with HH and obesity [23, 24]. Leptin has a central effect on the release of neurotransmitters, such as neuropeptide Y, which are believed to subsequently regulate GnRH secretion [1]. Patients with mutations in prohormone convertase 1 (PC1) also present with obesity and HH, as well as hypocortisolaemia and hypoinsulinaemia [25]. PC1 is an endopeptidase required for post-translational processing of some prohormones and neuropeptides.
GPR54 and Pubertal Onset
In 2003, loss-of-function mutations of the gene encoding GPR54 (G protein-coupled receptor 54) were shown to result in isolated HH [26, 27]. Kisspeptin-54, a 54-amino acid peptide derived from the KiSS1 gene product, is the natural ligand for GPR54, and significantly, a KiSS1 mutation associated with isolated HH has also been reported [28]. Shorter C-terminal derivatives of human kisspeptin-54, designated kisspeptin-14, -13, and -10, have similar high-affinity binding to GPR54 [29]. Intracerebroventricular kisspeptin-10 administration induces a dramatic release of GnRH in sheep [30], and intracerebroventricular kisspeptin-10 administered to primates results in an immediate gonadotrophin surge [31]. Gpr54 knockout mice [27] fail to secrete FSH and LH in response to exogenous murine kisspeptin-15, despite having anatomically normal hypothalamic GnRH neurons, indicating that the hypogonadism results from abnormalities of GnRH neuronal secretion [30]. Kisspeptin-induced GPR54 signalling is thus a major regulatory control point for GnRH release, and may play a determining role in pubertal onset. Recently, intravenous kisspeptin-54 was shown to also stimulate LH, FSH, and testosterone secretion in human male volunteers [32]. However, when infused continuously into male juvenile rhesus monkeys, human kisspeptin-10 appears to desensitize/downregulate Gpr54-induced GnRH release, monitored indirectly by gonadotrophin release [33]. These findings have potential therapeutic implications for a range of human reproductive conditions.
Kallmann’s Syndrome
Introduction
Aureliano Maestre de San Juan (1856) [34] first reported the autopsy finding of a hypogonadal man with small testes and absent olfactory bulbs. Franz Josef Kallmann (1944) [35] subsequently demonstrated the hereditable form of HH with anosmia in three different families. KS is a heterogeneous developmental genetic disorder affecting about 1 in 8,000 males and 1 in 40,000 females, the majority being sporadic [36]. Although HH with anosmia or hyposmia were previously thought to identify a distinct subgroup of HH (KS), recent findings cast doubt on this classification, and suggest that anosmia/normosmia and HH form a phenotypic continuum that can result from mutations in the same gene [37, 38]. Other clinical features associated with KS may include unilateral renal agenesis, bimanual synkinesia (upper body mirror movements), dental agenesis, and cleft palate.
In KS, anosmia and HH result from olfactory bulb (OB) dysgenesis and hypothalamic GnRH deficiency respectively [39]. In most vertebrates, olfactory and GnRH neurons both originate from the nasal compartment. Olfactory axons migrate and establish contact with the OB anlage, a prerequisite for OB morphogenesis [40]. There are two waves of GnRH neuronal migration into the brain: one precedes OB formation, the other occurring in association with OB formation (fig. 2) [5]. In this later wave of migration, GnRH-secreting neurons migrate into the hypothalamus along a nasal mesenchymal scaffold of olfactory, vomeronasal and terminal nerves, expressing neural cell adhesion molecule along their central processes, prior to dispersing in the mediobasal part of the anterior hypothalamus [41]. In 1989, Schwanzel-Fukuda et al. [42] described a human foetus with X-linked KS (XKS) whose GnRH neurons terminated in a tangle beneath the forebrain on the dorsal surface of the cribriform plate; this demonstrated that the HH in KS resulted from a GnRH neuronal migratory defect. Since the initial differentiation and migration of olfactory and GnRH neurons appeared normal, the developmental defects seen in XKS patients were proposed to result from abnormalities in subsequent axonal elongation, pathfinding, and/or terminal differentiation [42, 43].
![Fig. 2. Schematic drawing depicting anosmin-1 (green star), FGFR1 (blue star), and GnRH (red circle) immunoreactivity in the olfactory system and rostral forebrain during human embryogenesis after 53–54 days (CS21). Red circles with blue stars at their centre represent GnRH cells that co-express FGFR1. F = Forebrain; OB = olfactory bulb; OE = olfactory epithelium; ON = olfactory nerve; TN = terminal nerve; NTg = terminal nerve ganglion cells, sc = sulcus circularis, LOT = lateral olfactory tract, gc = granule cells, m = meninges, Lv = lateral ventricle, ne = neuroepithelium [5, 43], and unpubl. data].](https://karger.silverchair-cdn.com/karger/content_public/journal/hrp/67/5/10.1159_000098156/4/m_000098156_f02.jpeg?Expires=1716310068&Signature=zU63jPmABELw7IjLmPfbeZnMTyYTP2J-cCUrpcEMpRranS-EwM1FZR0bHujKWl8Y9pXRrwUdWXc4sM-qt9KejxGaNC5hnO5EWsX~7cQSDx70spftmaWkiaLSi833HzHBNosC1A5huKh6pGJRN1BnoKP8R-S5v7Na1cJ1U0kXHoS7bKoseYoQnRFHUnMxOztFc0-8PC-nXImGvcHFDHFVg2IafiAX-Ug4A4tPpKiR3Vsy1ImmMHgng62I6ye~gQadWhRrWpQB-SbNadMCIRrzVwZBseBIz9WVyVpDwS45V6A8MZvLoP4KvWk0215uWcKIpjPlBb8RzcOn2lVFbpzR3g__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Schematic drawing depicting anosmin-1 (green star), FGFR1 (blue star), and GnRH (red circle) immunoreactivity in the olfactory system and rostral forebrain during human embryogenesis after 53–54 days (CS21). Red circles with blue stars at their centre represent GnRH cells that co-express FGFR1. F = Forebrain; OB = olfactory bulb; OE = olfactory epithelium; ON = olfactory nerve; TN = terminal nerve; NTg = terminal nerve ganglion cells, sc = sulcus circularis, LOT = lateral olfactory tract, gc = granule cells, m = meninges, Lv = lateral ventricle, ne = neuroepithelium [5, 43], and unpubl. data].
Schematic drawing depicting anosmin-1 (green star), FGFR1 (blue star), and GnRH (red circle) immunoreactivity in the olfactory system and rostral forebrain during human embryogenesis after 53–54 days (CS21). Red circles with blue stars at their centre represent GnRH cells that co-express FGFR1. F = Forebrain; OB = olfactory bulb; OE = olfactory epithelium; ON = olfactory nerve; TN = terminal nerve; NTg = terminal nerve ganglion cells, sc = sulcus circularis, LOT = lateral olfactory tract, gc = granule cells, m = meninges, Lv = lateral ventricle, ne = neuroepithelium [5, 43], and unpubl. data].
Anosmin-1 and XKS
KAL1, the gene responsible for XKS, was cloned independently by two groups in 1991 [44, 45]. Located on Xp22.3, it has a non-functional Y chromosome homologue. KAL1 orthologues are present in chicken, zebrafish, fruitfly, and the nematode worm Caenorhabditis elegans [39]. KAL1 mutations only account for a small minority of KS cases (14% of familial and 11% of sporadic cases) [46]. The phenotype associated with KAL1 mutations varies significantly, even amongst monozygotic twins sharing the same mutation [47], emphasizing the likely role of modifier genes, epigenetic factors and environmental factors in the penetrance of KAL1 mutations.
KAL1 encodes an approximately 100-kDa extracellular-matrix glycoprotein [48], anosmin-1, which shares homology with neural cell adhesion molecule [44]. During development, anosmin-1 expression is restricted to basement membranes and interstitial matrices of discrete embryonic areas, including the developing olfactory bulb, retina, and kidney [49, 50], some correlating with the distribution of clinical significant abnormalities in XKS patients. Anosmin-1 is a multi-domain protein consisting of an N-terminal cysteine-rich region (Cys box), followed by a whey acidic protein-like (WAP) four disulphide core motif, four tandem fibronectin type III (FnIII) domains and a C-terminal histidine-rich region (fig. 3). The Cys box contains ten cysteine residues, resembling the cysteine-rich region of the insulin-like growth factor receptor [51]. The eight cysteines of the WAP domain form four disulphide bonds, highly conserved throughout the serine protease inhibitors of the WAP protein family, including elafin, SLPI and PS20 [52]. Large positively charged basic regions on the FnIII domains, particularly the first, were shown by surface plasmon resonance to be essential for high-affinity dose-dependent binding to negatively charged heparan sulphate (HS) [52]. We recently used both small angle X-ray scattering and analytical ultracentrifugation to characterize the solution structure of recombinant anosmin-1. Data generated were interpreted using constrained homology modelling to visualize the three-dimensional construction of anosmin-1; this showed that the six domains of anosmin-1 were extended with flexible inter-domain linkers, suggesting it may act as a platform for coordinate interaction with HS and its other biomacromolecular ligands [51].

Schematic domain-structure diagram of anosmin-1 (drawn to relative scale). SP = Signal peptide; Cys box = cysteine-rich region; WAP = whey acidic protein-like four disulphide core motif; FnIII-1, FnIII-2, FnIII-3, FnIII-4 = fibronectin type III domains; H = histidine-rich region.
Schematic domain-structure diagram of anosmin-1 (drawn to relative scale). SP = Signal peptide; Cys box = cysteine-rich region; WAP = whey acidic protein-like four disulphide core motif; FnIII-1, FnIII-2, FnIII-3, FnIII-4 = fibronectin type III domains; H = histidine-rich region.
Although anosmin-1 is highly conserved (especially the WAP and first FNIII domain) across many species, the rodent homologue has remained elusive, frustrating attempts to develop a mouse XKS model. However, one group has shown that anti-human anosmin-1 antibodies cross-react with a 100-kDa protein in cerebellum and OB extracts from both the rat and mouse, indicating that a rodent anosmin-1 orthologue might exist [53]. Recently, knockdown of a zebrafish KAL1 orthologue was shown to result in the specific loss of hypothalamic GnRH cells, while not affecting GnRH cells within the midbrain and terminal nerve [54]. Similarly, in the medaka fish model, knockdown of a KAL1 orthologue disrupted forebrain GnRH neuronal migration [55].
In vitro, Cariboni et al. [56] have shown that human anosmin-1 stimulates migratory activity in immortalized rodent GnRH-producing neurons, whereas Soussi-Yanicostas et al. [57] have demonstrated that anosmin-1 modulates neurite outgrowth in a cell-type specific manner. This same group subsequently showed that anosmin-1 stimulated collateral branch formation from rat OB output neurons (the mitral and tufted cells) [53]. However, previous observations suggest that olfactory defects seen in KS patients result from earlier abnormalities in olfactory system development; specifically, migration of sensory neuronal axons towards, and connection with, the OB anlage in the developing forebrain [42]. In this model, anosmin-1 expressed in the presumptive OB area is hypothesized to attract olfactory sensory neuronal axons towards the forebrain during the latter stages of their trajectory. In the absence of olfactory nerve synaptogenesis with the OB anlage, GnRH neurons are denied a navigational pathway towards the forebrain, explaining the deficiency of hypothalamic GnRH-secreting neurons in KS [53].
The Role of HSPG
During development, the ability of anosmin-1 to confer GnRH neurons with cell-specific chemotactic responsivity, as well as branch-promoting and guidance functions in olfactory neurons, is dependent on heparan sulphate proteoglycan (HSPG) interactions. In fact, HSPGs are a critical component of the ECM, playing a vital role in neuronal navigation during CNS development [58].
HSPGs are cell-surface or secreted proteins containing the glycosaminoglycan HS. During HS chain biosynthesis, alternating glucuronic acid (GlcA) and N-acetylglucosamine (GlcNAc) subunits are extended from the core tetrasaccharide attached to a serine residue on the protein. Some of the GlcNAc residues are then modified by N-deacetylation/N-sulphation to form N-sulphated glucosamine (GlcNS) regions (NS domains) which are interspersed within the unmodified GlcNAc regions (NA domains). The polymer is then further modified by epimerization of GlcA to iduronic acid (IdoA) and by three types of sulphation; the 2-O-sulphation of IdoA (or more rarely GlcA) to form IdoA(2S) or GlcA(2S); the 6-O-sulphation of GlcNS residues to form GlcNS(6S); and the 3-O-sulphation of GlcNS(6S) to form GlcNS(3S,6S). All modification reactions are incomplete in vivo, generating many HS chain variants with distinct, highly variable, domains of charge density, thereby altering their molecular binding specificities.
In C. elegans, particular HS modifications are required for anosmin-1 activity in certain cell types. ‘AIY interneurons’ are a subclass of C. elegans neurons which receive synaptic input from olfactory neurons. The C. elegans orthologue of anosmin-1 (ceKal1) induces a specific axonal branching phenotype in these cells, which was abolished in worms lacking 6-O-sulphotransferase (hst-6) or C5-epimerase (hse-5), but not in those lacking 2-O-sulphotransferase (hst-2). However, hypodermal defects induced by ceKal1 overexpression were suppressed only in those worms lacking hse-5, but not by those lacking hst-6 or hst-2. It was therefore proposed that anosmin-1 function requires distinct HS modifications in different developmental contexts [59, 60]. Significantly, HS saccharide alterations were also shown to affect binding specificity of ligand-receptor interactions of the fibroblast growth factor receptor 1 (FGFR1) [61], which has since been identified as KAL2 [37].
FGFR1 and Autosomal Dominant KS
In 2003, Dode et al. [37] described 2 patients with different contiguous gene syndromes that both included KS in their pathology. The overlap of the deletion in each of these patients, located at chromosome 8p11.2–p12, contained three previously characterized genes, including FGFR1, hypothesized to be the most likely KS-associated locus. Loss-of-function mutations in FGFR1 were subsequently shown to be associated with some cases of autosomal dominant KS (KAL2) (table 1, fig. 4). By contrast, gain-of-function mutations in FGFR1 had previously been shown to result in craniosynostosis. As with KAL1 mutations, penetrance of KAL2 mutations varies considerably, even within the same kindred. Thus, Pitteloud et al. [38] have described 3 subjects from the same family sharing an identical tyrosine kinase domain FGFR1 mutation, but each with a different phenotype. The familial kindred comprised a male proband with KS (who later recovered from his HH), whose mother had delayed puberty and whose maternal grandfather had isolated anosmia.
A summary of the reported loss-of-function FGFR1 mutations found in patients with autosomal dominant KS


Schematic domain-structure diagram of FGFR1. The protein (top) and the corresponding exons (bottom) are shown. The extracellular regions of FGFR1 that have a role in autoinhibition or ligand binding are indicated (see main text for further discussion). SP = Signal peptide; D1, D2, and D3 = the three immunoglobulin-like domains; AB = acid box; HBS = HS binding site; IIIb/IIIc refers to two major splice isoforms; TM = transmembrane helix, and PTK refers to the intracellular protein tyrosine kinase domain. Missense mutations are indicated by arrows; all other mutations are represented by stars.
Schematic domain-structure diagram of FGFR1. The protein (top) and the corresponding exons (bottom) are shown. The extracellular regions of FGFR1 that have a role in autoinhibition or ligand binding are indicated (see main text for further discussion). SP = Signal peptide; D1, D2, and D3 = the three immunoglobulin-like domains; AB = acid box; HBS = HS binding site; IIIb/IIIc refers to two major splice isoforms; TM = transmembrane helix, and PTK refers to the intracellular protein tyrosine kinase domain. Missense mutations are indicated by arrows; all other mutations are represented by stars.
FGFR1 Structure and Autoinhibition Control
FGFR1 signalling has been known to play a wide-ranging role during embryogenesis, homeostasis and wound healing. In humans, there are four known members of the FGFR family of receptor tyrosine kinases (FGFR1, FGFR2, FGFR3, and FGFR4), which specifically bind to particular members of the 22 FGF ligands, in different cellular contexts. As well as requiring two FGF ligands, HS is also essential for FGF receptor dimerization and activation. FGFRs usually consist of an extracellular region of three immunoglobulin (Ig)-like domains (D1, D2, and D3), a single transmembrane helix, and a cytoplasmic tyrosine kinase domain (fig. 4). The D1-D2 linker region contains a stretch of negatively charged amino acids (the acid box), and there is a HS-binding site (HBS) within the first half of D2. Alternative splice variants exist for FGFRs 1, 2, and 3. The common ‘IIIa exon’ (exon 7) that encodes the first half of D3 can be spliced to either exon 8A or 8B, resulting in the ‘FGFR1 IIIb’ and ‘FGFR1 IIIc’ isoforms. When neither 8A nor 8B is used, the result is a soluble ‘FGFR1 IIIa’ variant []reviewed in [62].
As with other receptor tyrosine kinases, FGFR1 plays a significant role in cell proliferation and differentiation. Its activity is therefore under tight regulation. According to a recently proposed ‘autoinhibition model’, in quiescent cells, the acidic box may bind to the positively charged HBS, consequently bringing the D1 domain into a position that will interfere with HS and FGF ligand binding to the D2-D3 regions, resulting in a closed, autoinhibited state. The presence of FGF, which has a higher affinity for the D2-D3 ligand binding sites, is hypothesized to open up the closed configuration, making the HBS accessible to HS (the obligatory cofactor for functional activation of the whole signalling complex). In quiescent cells, ‘closed’ (autoinhibited) FGFR1 is in equilibrium with the ‘open’ (active) FGFR1 configuration. Therefore, upon binding of HS and FGF, the equilibrium shifts towards the ‘open’ state and the FGFR1 dimerizes and becomes fully active. KS patients, whose loss-of-function mutations map to D1 or the acid box, may have a more autoinhibited FGFR1, thus disturbing the equilibrium and resulting in FGFR1 signalling insufficiency [63].
FGFR1 and Olfactory GnRH System Development
Hebert et al. [64] showed that the targeted abolition of Fgfr1 expression in the developing rodent embryonic telencephalon resulted in OB aplasia, confirming the primacy of FGFR1 signalling for OB morphogenesis. Furthermore, FGF8 was shown to be a particularly important ligand for olfactory development; thus mice with a partial loss of Fgf8 have a small telencephalon without an OB [65]. FGF2 is co-expressed with FGFR1 in the nasal epithelium, and may therefore represent a further FGFR1 ligand for signalling during olfactory GnRH system development [66].
Specific expression of a dominant-negative Fgfr1 (dnFgfr) in GnRH neurons in mice resulted in a 30% decrease in GnRH neurons of the forebrain [67] and a significant reduction in GnRH axonal projections towards the median eminence [68]. dnFgfr lacks the intracellular tyrosine kinase domain, and upon binding ligand, forms a non-functional heterodimer with wild-type Fgfr1, Fgfr2, and Fgfr3, thus blocking subsequent downstream Fgfr signalling [67].
Anosmin-1 Enhances FGFR1 Signalling
Anosmin-1 and FGFR1 are found in the olfactory placode of human embryos as early as 4.5 weeks, and later, at 8 weeks, are both present at the terminal nerve region, known to be part of the ‘scaffold’ for the migratory path of GnRH-expressing neurons towards the hypothalamus (fig. 5). The close proximity of anosmin-1 with FGFR1 at these stages supported the possibility of their putative in vivo interaction during olfactory GnRH system development. We have recently demonstrated that anosmin-1 is indeed directly involved in FGFR1 signalling, thus providing a link between X-linked and autosomal dominant KS. In human embryonic GnRH olfactory neuroblasts, anosmin-1 induced neurite outgrowth and cytoskeletal changes through an FGFR1-dependent mechanism. Anosmin-1 enhanced FGF2 signalling specifically through the FGFR1 IIIc isoform, in a HS-dependent manner, although further work needs to be carried out to investigate a potential role for anosmin-1 in regulating the IIIb isoform [43]. These dose-dependent effects of anosmin-1 on FGFR1 signalling help to explain the higher prevalence of KS in males. KAL1 partially escapes X-inactivation in females which results in them having higher levels of anosmin-1 dosage compared to males; this could compensate for deficient FGFR1 signalling in heterozygous females [37].
![Fig. 5. Expression of anosmin-1, FGFR1, and GnRH in 4.5- and 8-week-old human embryo sections. A Immunohistofluorescence of a 4.5-week-old human embryo OP showing FGFR1 (FITC labelling) and GnRH (Texas Red labelling) protein distribution. Yellow arrowheads indicate sites of co-expression of these proteins in the medial region of the OP. B The adjacent serial section, 7 µm down from A, showing FGFR1 (FITC labelling) and anosmin-1 (Texas Red labelling) distribution in the OP. C Double immunohistochemistry of an 8-week-old embryo section, showing anosmin-1 (brown DAB precipitates; black arrowheads) and FGFR1 (crimson Fast Red precipitates; white arrowheads) in the TN. D Double immunohistochemistry on an adjacent serial section of C, showing anosmin-1 (brown DAB precipitates; black arrowheads) and GnRH (crimson Fast Red precipitates; white arrowheads) in the TN. Scale bar: A , B = 100 µm; C, D =15 µm. r = Rostral; OP = olfactory placode; TN = terminal nerve. All sections are shown in sagittal orientation [43] (copyright 2004 by the Society for Neuroscience).](https://karger.silverchair-cdn.com/karger/content_public/journal/hrp/67/5/10.1159_000098156/4/m_000098156_f05.jpeg?Expires=1716310068&Signature=V0wKgAO1q-Q7JHlme5Q6yqCJL2yRVPRiVohhv4V5dUSt3pYgnyQTw3AqjCGdssfoUQ3MdrwmgkcXAcM-~bphvqYXbYfeobaCZzhNU7qoq9esAjPrVcu6fl4GfI4v22ELcpEoeo7OKvDMEQmlrpiCuilvEGLFdyp8pmzbcY0SAmIMBxQG4o3ga8yWRNOYpYb8pJs7kM7MjkYOo25ohMjq7Ex4Ws5Z7UJa7bm5kW8fvXaAQ805RhvbvF~6mjk2NwmYuHrdsJe5v4OQJHI5ELNecGM~VoTi6WdKVrVwLLLGoahP6DCZSkFZhnJ3O-2XDLK3h-i3gYRyJqI7U9LtcfxvdA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Expression of anosmin-1, FGFR1, and GnRH in 4.5- and 8-week-old human embryo sections. A Immunohistofluorescence of a 4.5-week-old human embryo OP showing FGFR1 (FITC labelling) and GnRH (Texas Red labelling) protein distribution. Yellow arrowheads indicate sites of co-expression of these proteins in the medial region of the OP. B The adjacent serial section, 7 µm down from A, showing FGFR1 (FITC labelling) and anosmin-1 (Texas Red labelling) distribution in the OP. C Double immunohistochemistry of an 8-week-old embryo section, showing anosmin-1 (brown DAB precipitates; black arrowheads) and FGFR1 (crimson Fast Red precipitates; white arrowheads) in the TN. D Double immunohistochemistry on an adjacent serial section of C, showing anosmin-1 (brown DAB precipitates; black arrowheads) and GnRH (crimson Fast Red precipitates; white arrowheads) in the TN. Scale bar: A , B = 100 µm; C, D =15 µm. r = Rostral; OP = olfactory placode; TN = terminal nerve. All sections are shown in sagittal orientation [43] (copyright 2004 by the Society for Neuroscience).
Expression of anosmin-1, FGFR1, and GnRH in 4.5- and 8-week-old human embryo sections. A Immunohistofluorescence of a 4.5-week-old human embryo OP showing FGFR1 (FITC labelling) and GnRH (Texas Red labelling) protein distribution. Yellow arrowheads indicate sites of co-expression of these proteins in the medial region of the OP. B The adjacent serial section, 7 µm down from A, showing FGFR1 (FITC labelling) and anosmin-1 (Texas Red labelling) distribution in the OP. C Double immunohistochemistry of an 8-week-old embryo section, showing anosmin-1 (brown DAB precipitates; black arrowheads) and FGFR1 (crimson Fast Red precipitates; white arrowheads) in the TN. D Double immunohistochemistry on an adjacent serial section of C, showing anosmin-1 (brown DAB precipitates; black arrowheads) and GnRH (crimson Fast Red precipitates; white arrowheads) in the TN. Scale bar: A , B = 100 µm; C, D =15 µm. r = Rostral; OP = olfactory placode; TN = terminal nerve. All sections are shown in sagittal orientation [43] (copyright 2004 by the Society for Neuroscience).
FGFR1-Independent Anosmin-1 Signalling?
We have also shown that anosmin-1 significantly enhances the amidolytic activity of urokinase-type plasminogen activator (uPA) in vitro. This is hypothesized to enhance the uPA-induced activation of the plasmin cascade at the cell surface and consequently result in proteolytic degradation of ECM components that subsequently release cell-cell and cell-ECM interactions that facilitate cell migration and neuronal synaptogenesis. Also, the anosmin-1-HS-uPA interaction was shown to induce cell proliferation in the PC-3 prostate carcinoma cell line, in an FGFR1-independent manner (as these cells lack this receptor) [52]. Furthermore, FGF2 induces uPA expression in mouse brain capillary endothelial cells, which suggests that there could also be a functional relationship between uPA and FGF signalling in vivo [69].
Other KS-Associated Loci?
Although the majority of KS cases are caused by mutations in hitherto unidentified genes, recent progress has led to the identification of three promising new candidates: CHD7, NELF, and PKR2.
CHD7 mutations are responsible for some cases of CHARGE syndrome [70], a developmental disorder defined by coloboma, congenital heart disease, choanal atresia, mental and growth retardation, genital hypoplasia, and ear malformations and/or deafness [71, 72]. Significantly, the defining clinical features of KS, HH with olfactory deficiency, are also present in patients with CHARGE syndrome [73]. In fact, some cases of KS had previously been reported with additional phenotypes including choanal atresia, mental retardation, and hearing loss [74, 75], thus demonstrating that there is considerable overlap between KS and CHARGE syndrome. CHD7 therefore represents a new locus to be investigated in suspected KS patients who do not have mutations in either KAL1 or FGFR1. The protein encoded by CHD7, chromodomain helicase DNA-binding protein-7, is a member of a superfamily of proteins that uniquely comprise two N-terminal chromodomains, an SNF2-like ATPase/helicase domain and a DNA-binding domain [76]. It is expressed in many foetal (and adult) tissues, including the developing brain, and is predicted to affect both chromatin structure and gene expression during early embryonic development [70].
Nelf, the mouse nasal embryonic LHRH (GnRH) factor gene, encodes a guidance molecule required for olfactory axonal outgrowth and GnRH neuronal migration in mice. The human orthologue, NELF, therefore seemed to be a likely candidate as a KS-associated locus. However, only one case of HH with a novel heterozygous mutation in this gene is known [77], and there has been no definitive link to KS so far.
Finally, a recent report showed that prokineticin receptor 2 (Pkr2) knockout mice have a phenotype which is very similar to that seen in KS patients [78]. PKR2 (and PKR1) is a G protein-coupled receptor, which is activated by the prokineticins, PK1 and PK2. In 2005, Ng et al. [79] demonstrated that PK2 signalling is essential for normal OB formation; mice with Pk2 deficiency have a marked reduction in OB size, and abnormal OB architecture. Matsumoto et al.[78] recently showed that Pkr2 knockout mice exhibit OB hypoplasia, which, significantly, is associated with severe atrophy of the reproductive system, including the testis, ovary, uterus, vagina, and mammary gland. These Pkr2 knockout mice have decreased plasma levels of testosterone and FSH, and immunohistochemical analysis revealed an absence of hypothalamic GnRH neurons. However, mutation analysis of human PKR2 will need to be carried out before it is known whether this gene is a bona fide KS-associated gene.
Conclusions
By identifying the genes that are mutated in HH patients, and deciphering the signalling pathways their encoded proteins are involved in, some of the complexities of the human reproductive axis are being gradually unravelled, both in terms of its development and the modulation of its physiology from birth to adulthood. These approaches may generate novel therapeutic targets for both HH and in the area of population control. An example of this would be the pharmacological manipulation of the GPR54 system, thereby modulating GnRH secretion, thus altering reproductive competency. The demonstration of a functional HSPG-dependent interaction between anosmin-1 and the FGFR1/FGF2 signalling complex has also identified a novel pathway for modulation of FGFR1 signalling. An understanding of the mechanism of this interaction may pave the way for identifying novel mechanisms for regulation of FGFR1 signalling and may have potential therapeutic implications for neuronal repair.
Anosmin-1, the first protein identified to be involved in the pathophysiology of KS, is highly conserved across many species, and has an important role in neuronal cell migration, guidance, branching, and possibly terminal differentiation. The discovery that anosmin-1 is a positive regulator of FGFR1 (the only other known KS-associated protein) illustrates the importance of the fine tuning of FGFR1 signalling during GnRH and olfactory neuronal development. Future work will involve determining the precise molecular mechanism of how anosmin-1 positively regulates FGFR1 signalling.
