

Abstract
Osteoimmunology is an interdisciplinary research field combining the exciting fields of osteology and immunology. An observation that contributed enormously to the emergence of osteoimmunology was the accelerated bone loss caused by inflammatory diseases such as rheumatoid arthritis. Receptor activator of nuclear factor ĸB ligand (RANKL), which is the main regulator of osteoclastogenesis, was found to be the primary culprit responsible for the enhanced activation of osteoclasts: activated T cells directly and indirectly increased the expression of RANKL, and thereby promoted osteoclastic activity. Excessive bone loss is not only present in inflammatory diseases but also in autoimmune diseases and cancer. Furthermore, there is accumulating evidence that the very prevalent skeletal disorder osteoporosis is associated with alterations in the immune system. Meanwhile, numerous connections have been discovered in osteoimmunology beyond merely the actions of RANKL. These include the importance of osteoblasts in the maintenance of the hematopoietic stem cell niche and in lymphocyte development as well as the functions of immune cells participating in osteoblast and osteoclast development. Furthermore, research is being done investigating cytokines, chemokines, transcription factors and co-stimulatory molecules which are shared by both systems. Research in osteoimmunology promises the discovery of new strategies and the development of innovative therapeutics to cure or alleviate bone loss in inflammatory and autoimmune diseases as well as in osteoporosis. This review gives an introduction to bone remodeling and the cells governing that process and summarizes the most recent discoveries in the interdisciplinary field of osteoimmunology. Furthermore, an alternative large animal model will be discussed and the pathophysiological alterations of the immune system in osteoporosis will be highlighted.
Introduction
The term ‘osteoimmunology’ was first used in the year 2000 as Aaron and Choi were highlighting the interdigitate communication between the immune and skeletal systems especially observed in autoimmune and other inflammatory diseases [1]. Major advances and discoveries in this interdisciplinary research field have lead to the revelation of molecular mechanisms as well as various cytokines and signaling transducers participating in the regulatory interplay between immune cells and bone cells. Furthermore, besides the arsenal of mutual signaling molecules, immune and bone cells also share a common site of origin, namely bone marrow. Due to the spatial proximity of the developing cells it is proposed that they influence each other not only after maturation and activation, as Kong and colleagues already observed in 1999 [2, 3], but also at the very beginning of their existence. Among others, Taichman and Emerson have described the important role of osteoblasts in the establishment of hematopoietic stem cell niches, as well as in the engraftment and maintenance of hematopoietic stem cells (HSC) in the bone marrow [4,5,6,7,8]. The development of bone cells has also been demonstrated to be supported by cells of the immune system; for instance, macrophages encourage osteoblastogenesis by the secretion of interleukin-18 [9], and T cells are capable of influencing osteoclastogenesis by the secretion of various cytokines such as interleukin-1, interleukin-6, interferon-γ or interleukin-4 [10, 11].
However, the most prominent osteoimmunological example arose from the observation of osteoclast-mediated bone loss in various inflammatory and autoimmune diseases such as rheumatoid arthritis, diabetes mellitus, lupus erythematosus, periodontal diseases and chronic viral infections (human immunodeficiency virus) [3,12,13,14,15,16]. Kong and colleagues were the first to notice that the osteoclast-inducing capacity of activated T cells in adjuvant arthritis was mediated through the RANKL/receptor activator of nuclear factor ĸB (RANK)/osteoprotegerin (OPG) axis. RANKL is a key regulator of osteoclastogenesis, thus contributing enormously to a process called bone remodeling. In this process, osteoclasts resorb old and damaged bone, which is then replaced by new bone material deposited by osteoblasts. As normal physiological bone remodeling is imperative for the maintenance of bone strength and integrity, imbalances either lead to increased or decreased bone mass the latter often being caused by inflammatory diseases.
Considering all of these intricate interrelationships between the immune and skeletal system and the high potential for developing innovative new therapeutic drugs targeting osteoclastogenesis-regulating cytokines, it is worth taking a closer look at the molecular mechanisms facilitating osteoclastogenesis and the environmental factors affecting that process.
Bone Function and Structure
Contrary to the general perception of bone being an inert, static material, it is a highly organized, living tissue and is the major constituent of the skeleton. The most prominent functions of bone are the protection of internal organs and the support of body structures. Beyond those functions bone additionally serves as an attachment site for muscles allowing locomotion and as an appropriate cavity for hematopoiesis in bone marrow. As a reservoir for inorganic ions, bone is responsible for the maintenance of calcium homeostasis and is able to rapidly mobilize mineral stores on metabolic demand.
Bone is composed of cells and extracellular matrix (ECM), the latter being further subdivided into an inorganic and organic part. The organic matrix is mainly constituted of type I collagen (approximately 95%), as well as other types of collagens, noncollagenous proteins and proteoglycans, whereas the inorganic matrix predominantly contains calcium and phosphorus, appearing as hydroxyapatite crystals ([3Ca3(PO4)2](OH)2) deposited into the collagenous matrix. This interdigitate organization confers rigidity and strength to the skeleton while maintaining a certain degree of elasticity. The major cells in bone are the osteoclasts, resorbing bone tissue, and osteoblasts (including osteocytes and bone lining cells), depositing bone tissue.
Osteoblasts and Osteoclasts
The main functions of osteoblasts are to synthesize the collagen-rich organic matrix and to provide optimal conditions for matrix mineralization by secreting numerous bone matrix proteins and matrix metalloproteinases (MMP) [17]. Furthermore, osteoblasts support hematopoiesis, notably osteoclastogenesis [18, 19]. Bone lining cells, one possible destiny of fully differentiated osteoblasts, are responsible for the initiation of bone remodeling by matrix degradation [20], whereas osteocytes, the other form of terminally differentiated osteoblasts, act as mechanosensors in bone tissue, thereby regulating bone mass and structure [21,22,23,24].
Osteoblasts, chondrocytes, adipocytes, stromal cells, myoblasts and tenocytes all originate from a common progenitor cell, the mesenchymal stem cell (MSC) [25,26,27]. The multipotent MSC undergoes several steps of commitment to give rise to progeny with more limited capacities until the differentiated end-stage cell is able to express particular functional markers and morphological traits. Core binding factor 1 (Cbfa1, also termed runt-related transcription factor 2, runx2) and the downstream factor osterix are crucial transcription factors for lineage commitment and osteoblast differentiation [28, 29]. The skeletons of Cbfa1 deficient mice only consist of cartilage, indicating its importance in osteoblast development [30, 31]. Recently, Jones et al. [32] identified the zinc-finger protein schnurri-3 (shn-3) which in association with WWP1 acts as a regulator of Cbfa1 expression by influencing cbfa1 degradation by ubiquitination.
Mature osteoblasts continue matrix deposition and start mineralization, expressing alkaline phosphatase and bone sialoprotein as well as osteocalcin and osteopontin [33]. Since osteoblasts produce a variety of proteins for bone matrix synthesis they are hallmarked by a prominent Golgi apparatus and rough endoplasmic reticulum. Osteoblasts turn into osteocytes after they have been surrounded by bone matrix. Osteocytes are poor in organelles, indicating other primary functions than matrix synthesis and mineralization. In fact, mature osteocytes alter their morphology by forming dendritic processes which enable them to communicate with other embedded osteocytes. These processes are believed to act as mechanosensors in bone tissue, which allow them to react to environmental changes [34,35,36,37]. Moreover, there is emerging evidence that apoptotic osteocytes increase the secretion of osteoclastogenic cytokines and thereby enhance bone resorption [38, 39]. Bone lining cells also arise from osteoblasts and are believed to be resting, inactive osteoblasts, covering the bone surface. As mentioned earlier, an important role in the initiation of bone remodeling has been assigned to them [40].
Osteoclasts are tissue-specific giant polykaryons derived from the monocyte/macrophage hematopoietic lineage and are the only cells capable of breaking down mineralized bone, dentine and calcified cartilage [41, 42]. The presence of RANKL and M-CSF (macrophage-colony-stimulating factor) are essential for the formation and fusion of multinucleated cells, expressing osteoclast-specific markers such as tartrate-resistant acid phosphatase (TRAP), cathepsin K, calcitonin receptor (CTR) and integrin receptors [43,44,45,46,47,48]. Via integrins, osteoclasts attach very tightly to the matrix, thereby creating an isolated lacuna (Howship’s lacuna) able to maintain an acidic environment necessary for matrix dissolution [49]. After attachment intracellular rearrangements lead to the polarization of the cell borders, whereas the sealing zone is adjacent to the basolateral domain and the ruffled border, respectively. At the opposite side of the ruffled border emerges the functional secretory domain (FSD) [50]. The ruffled border and the FSD are connected to each other via microtubules on which exocytotic vesicle traffic has been observed [51], suggesting the secretion of resorbed material into the extracellular space. In addition to the development of distinct membrane domains, the cytoskeleton undergoes organizational changes creating a dense actin ring in osteoclasts preparing for resorption.
The resorption of bone matrix takes place in the resorption lacuna [52]. The ruffled border is formed by the fusion of cytoplasmic acidic vacuoles, thereby releasing acid into the resorption lacuna and initiating rapid dissolution of the hydroxyapatite crystals [53]. Additionally, ATPases located in the ruffled border transport protons into the Howship’s lacuna. The protons are supplied by the reaction of water and carbon dioxide catalyzed by the enzyme carbonic anhydrase II resulting in the formation of protons and HCO3–. Whereas the protons are pumped into the resorption lacuna HCO3– is transported into the extracellular space via HCO3/Cl exchanger. The imported chloride ions are also pumped into the resorption lacuna to form hydrochloric acid capable of dissolving the mineralized matrix. The organic matrix is degraded by various enzymes, including tartrate-resistant acid phosphatase (TRAP), cathepsin K and matrix metalloproteinase 9 (MMP-9) [20,54,55,56].
Bone Remodeling
Continuously changing functional demands require permanent adaption of the bone structure and microarchitecture. Wolff [57 ]has observed this principle of functional adaptation already over 100 years ago. The process of where ‘form follows function’ [58] consists of two activities, namely, bone formation and bone resorption. While these processes are locally separated in modeling [59, 60], bone remodeling is characterized by the spatial and temporal coupling of bone formation by osteoblasts and bone resorption by osteoclasts [61]. Approximately 5–25% of bone surface is undergoing bone remodeling [62, 63], thereby restoring microdamages and ensuring mechanical integrity as well as regulating the release of calcium and phosphorus.
Bone remodeling involves four main processes: activation, resorption, reversal and formation [64]. The remodeling cycle is initiated by the activation of the quiescent bone surface, which is covered with bone lining cells [65, 66]. Osteoclast precursor cells are recruited to the activated surface and fuse to form mature, bone resorbing osteoclasts. The osteoclasts attach to the surface, dissolve the inorganic matrix by creating an acidic microenvironment, and degrade the organic matrix with specific enzymes. As bone resorption subsides and resorption pits remain, osteoclasts disappear and mononuclear cells prepare the surface for bone formation. The bone remodeling cycle is finished with the synthesis and deposition of bone matrix by osteoblasts, and bone lining cells building a canopy covering the surface, keeping the material dormant until the next cycle.
Regulation of Bone Remodeling
Prostaglandins, Leukotrienes and Hormones
Bone formation and resorption are under the subtle control of various local and systemic factors. Besides the multiple cytokines that participate in the regulatory system of bone homeostasis prostaglandins, leukotrienes and hormones additionally interact with bone cells, thereby affecting bone remodeling processes.
Prostaglandins and leukotrienes are metabolites of arachidonic acid and have stimulatory as well as inhibitory effects on bone [67]. In the presence of factors stimulating bone resorption, prostaglandin E2 (PGE2) is produced by the cyclo-oxygenase 2 (COX2)-mediated conversion of arachidonic acid in osteoblasts. Recently, Ha et al. [68] were able to demonstrate an inhibition of osteoclastogenesis primarily by the reduction of IL-1-induced COX2 activity and PGE2 production and only marginally by the suppression of RANKL by α-lipoic acid [69,70,71]. Additionally, leukotriene B4 was also shown to enhance osteoclastogenesis in a RANKL-independent manner [72,73,74].
The peptide hormone parathyroid hormone (PTH) is one of the most important regulators of calcium ion homeostasis [75, 76]. In response to low blood calcium levels, PTH is secreted into the circulation and acts on kidney, bone and intestine to maintain blood calcium concentrations. In bone, PTH upregulates the production of interleukin-6 and RANKL by osteoblasts, thereby facilitating the differentiation, activation and survival of osteoclasts [77,78,79,80,81,82]. Thus, PTH as well as PTHrP (PTH-related protein) promote bone resorption and consequently the release of calcium [83, 84].
The active hormonal form of vitamin D, calcitriol (1α,25-dihydroxyvitamin D3), is essential for the development and maintenance of the mineralized skeleton, as demonstrated in various studies using vitamin D receptor or 1α(OH)ase knock-out mice [85,86,87]. The impaired mineralization was normalized when a high-calcium, high-phosphate and high-lactose diet (rescue diet) was administered. However, Panda and colleagues were able to show that the administration of only 1,25(OH)2D3 to 1α(OH)ase knock-out mice was not sufficient to normalize the impaired mineralization if hypocalcemia was not corrected [85]. Furthermore, vitamin D-deficient mice showed an increase in osteoblast number, bone formation and bone volume as well as increased serum alkaline phosphatase levels. Additionally, osteoclast numbers were decreased due to a decreased production of RANKL and an enhanced production of OPG [88].
Concerning bone homeostasis, estrogen and androgens are the most intensively investigated sex steroids and, in contrast to PTH and 1,25-dihydroxyvitamin D3, enhance bone formation and inhibit bone resorption [89,90,91,92,93]. Estrogen as well as testosterone deficiency inevitably lead to an increased rate of bone turn-over, which has been demonstrated by Jilka et al.[ 94], who observed simultaneous increases in osteoclastic precursors as well as early osteoblastic precursors. Additionally, estrogen deficiency results in a net loss of bone as a consequence of an increased production of RANKL and a decreased production of OPG in osteoblastic cells as well as the enhancement of the secretion of pro-inflammatory and pro-resorptive cytokines in lymphocytes such as IL-1, IL-6 and tumor necrosis factor-α (TNF-α) [95,96,97,98,99,100]. Moreover, the bone-protective effect of estrogen has been demonstrated to be mediated by transforming growth factor-β (TGF-β), inducing apoptosis in osteoclasts [101,102,103]. In two randomized controlled trials from the Women’s Health Initiative, hormone replacement therapy (HRT) had been shown to decrease the incidence of major osteoporotic fractures [227, 228]. However, serious side effects such as cardiovascular disease and cancer have occurred and therefore other medications are used nowadays in the treatment of osteoporosis. Raloxifene is a selective estrogen receptor modulator (SERM) and is approved for the treatment of osteoporosis. Like estrogen, SERMs are known to mediate their effects through the estrogen receptor. While estrogen binds equally strongly to alpha and beta receptors, raloxifene preferentially binds to the alpha receptor. This specificity to a certain estrogen receptor allows a higher affinity to bone, and therefore the side effects of SERMs are less pronounced than those of HRT [229].
The RANKL/OPG/RANK Network
The discovery of RANKL and its receptors RANK and OPG has highlighted the molecular processes in osteoclastogenesis, raising the possibility to inhibit the development of osteoclasts, rescuing bone from exorbitant resorption. In 1997, Simonet et al.[ 104] discovered a protein which exposed an osteopetrotic phenotype when overexpressed in transgenic mice. Investigating further, they found that this protein was secreted by preosteoblasts/stromal cells and was capable to inhibit osteoclast development and activation. Due to its bone-protective effects they named it osteoprotegerin. OPG belongs to the TNF receptor superfamily, however lacking a transmembrane and cytoplasmic domain. OPG is expressed in a variety of tissues, including lung, heart, kidney, liver, stomach, intestine, brain, spinal cord, thyroid gland, smooth muscle tissue and bone, indicating multiple possible functions [105,106,107,108,109,110]. The most prominent role of OPG has been assigned to bone protection; however, recent investigations have also proposed important functions of OPG in endothelial cell survival [111, 112] and vascular calcification [113,114,115].
After the identification of OPG followed the discovery of RANKL, which possesses not only a huge repertoire of names (TRANCE: TNF-related activation-induced cytokine; ODF: osteoclast differentiating factor; OPGL: osteoprotegerin ligand; TNFSF11: TNF superfamily member 11) but also many faces regarding its structure, function and appearance in tissues. The names originated from the four discoverers, each one having used different approaches to identify the protein. They searched either for a ligand for OPG [106], screened for apoptosis-regulating genes in T cell hybridomas [116], or found RANKL to induce osteoclastogenesis [117] and enhance the life-span of dendritic cells [118]. Kartsogiannis et al. [119] detected RANKL protein and mRNA expression in a variety of tissues, including bone, brain, heart, kidney, liver, lung, intestine, skeletal muscle, mammary tissue, placenta, spleen, thymus and testis [reviewed in [120]. This extensive distribution of RANKL throughout the body already indicates its multiple functions, whereas the most important one is dedicated to the induction of osteoclastogenesis, thus, the regulation of bone remodeling. RANKL knock-out mice reveal a severe osteopetrotic phenotype due to the absence of osteoclasts. Furthermore, defects in tooth eruption, lymph node genesis, mammary gland and lymphocyte development were reported as well as disturbances in T cell/dendritic cell interactions [121, 122]. Recently, Jones et al.[ 123] reported the observation of RANKL triggering cell migration of cancer cells expressing the receptor RANK indicating a role for RANKL as a chemo-attractant for cancer cells.
RANKL is a member of the TNF superfamily and is mainly expressed in preosteoblasts/stromal cells as well as on activated T cells [106, 117, 118, 124] and is closely related to the TNF-related apoptosis-inducing ligand TRAIL (homology ∼30%) and FasL (homology ∼20%). The human RANKL gene has been localized to chromosome 13q14 and encodes for three isoforms: RANKL1 and RANKL2 are type II transmembrane proteins, whereas RANKL2 encodes for a shorter intracellular domain. RANKL3 is a soluble protein, partially produced by the cleavage of the membrane-bound form by TACE (TNFα-converting enzyme, a metalloprotease) or other MMPs and by the direct transcription of the alternatively spliced RANKL-encoding gene [125,126,127,128,129].
The expression of OPG and RANKL is highly inducible by various systemic and local factors. Among others, estrogens, bone morphogenetic protein (BMP) 2, INF-γ and TGF-β positively regulate OPG, whereas PTH, 1,25(OH)2 vitamin D3, glucocorticoids, prostaglandin E2, IL-6, IL-8 and IL-11 enhance the expression of RANKL [reviewed in [130]].
The third participant in the bone remodeling regulatory system is RANK (receptor activator of nuclear factor ĸB) and belongs to the TNFR superfamily like OPG [118]. RANK represents a type I transmembrane protein and is expressed in tissues as ubiquitously as RANKL, although most commonly found in osteoclasts and dendritic cells [131]. RANK-deficient mice show similar phenotypes to those of RANKL knock-out mice, including tooth eruptions, osteopetrosis and missing lymph nodes [132].
The RANK signaling cascade (fig. 1) is initiated when RANKL binds to the extracellular domain of RANK which passes the signal along to TRAF6 (TNF receptor-associated factor 6) [133,134,135,136,137,138]. By transfecting RANK deficient cells in RANK mutants that are incapable of binding either TRAF 1, 2, 3, 5, or 6 Armstrong et al.[ 134] were able to demonstrate that predominantly TRAF6 was essential for the induction of osteoclastogenesis. TRAF6 has various downstream mediators which control the expression of osteoclast-specific genes during differentiation and activation of osteoclasts. The two most investigated pathways are the activation of the transcription factors NF-ĸB and AP-1 (activated protein 1). Targeted disruptions of the p50/p52 component of NF-ĸB and the c-fos component of AP-1 resulted in impaired osteoclastogenesis and revealed an osteopetrotic phenotype [139,140,141]. AP-1 is activated by signaling cascades mediated by JNK (c-Jun N-terminal kinase) whereas the phosphorylation of the inhibitor of NF-ĸB kinase (IKK) leads to the activation of NF-ĸB [142,143,144,145,146,147]. Other cascades of mitogen-activated protein kinases such as the TGF-β-inducible kinase TAK1 and the p38 stress kinase have also been described to participate in RANK signal transduction [148,149,150]. p38 is activated via the phosphorylation by MKK6 and in turn activates the transcriptional regulator mi/Mitf, which is responsible for the transcriptional control of genes encoding for the osteoclast-specific enzymes TRAP and cathepsin K [151, 152]. ERK (extracellular signal-related kinase) is a downstream target of MEK1 and acts as a negative regulator of osteoclastogenesis for ERK inhibitors have shown to accelerate RANKL-induced osteoclastogenesis [153]. The serine/threonine kinase Akt and the phosphatidylinositol-3-OH kinase (PI(3)K) are downstream elements of src and are known to mediate cell survival, motility and cytoskeletal rearrangements by the activation of the MEK/ERK and Akt/NFĸB pathways [121,154]. Recently, AFX/FOXO4 was shown to be the key downstream mediator activated by Akt/PKB to modulate osteoclast survival [155].
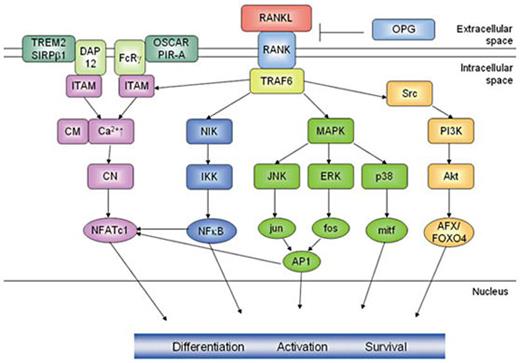
RANK signaling in osteoclasts. The RANK signaling cascade is initiated upon the binding of RANKL to the extracellular domain of RANK which passes the signal along to TRAF6. The activation of TRAF6 initiates pathways leading to the activation of the transcription factors NFAT, NFkB, the MAP kinase mediators jun, fos and p38 as well as the down-stream targets of Akt AFX/FOXO4, which contribute to osteoclast differentiation, activation and survival. The ITAM-containing co-stimulatory molecules DAP12 and FcRγ, respectively, initiate Ca2+-signaling leading to the activation of NFATc1. This schematic representation only focuses on the most important pathways, not illuminating further interactions of the signaling mediators.
RANK signaling in osteoclasts. The RANK signaling cascade is initiated upon the binding of RANKL to the extracellular domain of RANK which passes the signal along to TRAF6. The activation of TRAF6 initiates pathways leading to the activation of the transcription factors NFAT, NFkB, the MAP kinase mediators jun, fos and p38 as well as the down-stream targets of Akt AFX/FOXO4, which contribute to osteoclast differentiation, activation and survival. The ITAM-containing co-stimulatory molecules DAP12 and FcRγ, respectively, initiate Ca2+-signaling leading to the activation of NFATc1. This schematic representation only focuses on the most important pathways, not illuminating further interactions of the signaling mediators.
However, on the level of transcription factors nuclear factor of activated T cells c1 (NFATc1) has been elected as the master regulator of osteoclastogenesis. Many downstream effectors of RANK such as NF-ĸB and AP-1 contribute to the activation of NFATc1. Furthermore, RANKL-induced Ca2+-signaling, mediated by immunoreceptor tyrosine-based activation motifs (ITAMs), has been shown to be indispensable for osteoclastogenesis, since mice deficient in the ITAM-containing adaptor molecules DAP (DNAX-activating protein) 12 and Fc common receptor γ chain (FcRγ) are severely osteopetrotic. After retroviral transfer of DAP12 the osteopetrotic phenotype was rescued. The association of paired immunoglobulin-like receptor A (PIR-A) and OSCAR to FcRγ and triggering receptor expressed on myeloid cells (TREM) 2 and signal-regulatory protein β1 (SIRPβ1) to DAP12 in osteoclast precursors is considered to act as a co-stimulatory signal for RANKL, since one signal by its own is not able to induce osteoclastogenesis [156,157,158].
Cytokines
Recent studies dealt with the effects cytokines have on the generation of osteoblasts and osteoclasts. It is known that IL-1α, IL-1β, IL-6 and other members of the gp130 cytokine family, IL-7 and TNF-α directly or indirectly promote osteoclastogenesis [159,160,161,162], whereas interferon-beta (IFN-β), IFN-γ, IL-3, IL-4, IL-10, IL-13, and IL-12 alone and in synergy with IL-18 [163,164,165,166,167,168], amongst others, inhibit osteoclast formation. TGF-β was found to both induce, via suppressor of cytokine signaling 3 (SOCS3) [163], and suppress osteoclastogenesis [169]. For a detailed list, see the review by Theolyre et al. [120].
Among the osteoclastogenesis-inhibiting cytokines, interferons have attracted increasing attention. Thus, IFN-γ, a cytokine produced by activated T cells, was identified to strongly suppress osteoclastogenesis by inhibiting RANKL signaling by downregulation of the transcription factor TRAF6 expression via Stat1. The same applies for IFN-β, which is especially interesting in that this cytokine is induced by RANKL via a second down-stream molecule, namely c-Fos, but at the same time acts as negative regulator of RANKL signaling by inhibiting c-Fos expression in terms of a negative feedback loop. This interference of interferons with osteoclast differentiation was reviewed in detail by Takayanagi et al. [170].
However, there is little detailed information on the cytokine production pattern of osteoblasts. IL-6 was shown to be produced by stromal cells/osteoblasts [171]. A number of growth factors and hormones are known to promote proliferation and differentiation of osteoblasts, such as TGF-β (which is also assumed to depress osteoblast differentiation) [172], parathyroid hormone, its locally produced homologue parathyroid hormone-related peptide (PTHrP), low-density lipoprotein receptor-related protein-5 (LRP-5) [173], and osteopontin [174]. In recent research in osteology, much attention has been attributed towards bone morphogenic proteins (BMPs). So far, in pigs BMP-6 and BMP-7 have been shown to increase osteogenic differentiationin vitro [175], and BMP-4, besides IGF-1 and TGF-β, was found to be expressed during distraction osteogenesis in a pig model [176]. For human mesenchymal stem cells (MSCs), among BMP-2, -4, -6, and -7, BMP-6 was found to be the most consistent and potent regulator of osteoblast differentiation. Addition of BMP-6 to MSCs in vitro leads to the upregulation of type I collagen, osteocalcin, and bone sialoprotein [177]. Production of IL-6 and RANKL by osteoblasts is promoted by PTH and TNF-α, but with markedly different kinetics. Whereas PTH induces only a rapid, but transient elevation of both cytokines, TNF-α leads to a biphasic increase of these cytokines, thus indicating the potent role of TNF-α in pathologic conditions [77].
Interactions of Bone Cells with Lymphocytes
There is general agreement that lymphocytes influence bone remodeling by exerting an impact on osteoclastogenesis (fig. 2). Thus, T cells are assumed to be responsible for bone loss which occurs as a consequence of a series of pathological conditions, for example systemic viral infections and chronic local bone and joint diseases, such as rheumatoid arthritis or inflammatory bowel disease [3, 178]. Concerning the type of impact that T cells exert on osteoclastogenesis results from in vitro and in vivo experiments differ to a high degree. The same is true for different lymphocyte subpopulations, i.e. data concerning the effect of CD4 and CD8 lymphocytes on osteoclastogenesis, are not consistent. On the one hand, data from literature suggest an inhibitory effect of T cells. In one in vitro study, 1α,25(OH)2D3-stimulated osteoclast-like cell formation was enhanced after lymphocyte depletion. This was attributed to increased PGE2 production and consecutive upregulated RANKL and downregulated OPG expression [179]. IFN-γ was found to be the modulatory factor, which is produced by activated (by anti-CD3ε) T cells, and which interferes with TRAF6, thus strongly inhibiting the RANKL-induced activation of NF-ĸB and JNK in vitro. Resting T cells were found to exert no effect on osteoclastogenesis in the cited reference. These results were confirmed by another experiment, demonstrating that activated T cells have no effect in co-culture with IFN- γR–/– BMMs (bone marrow-derived monocyte/macrophage precursor cells) stimulated by RANKL [11]. In contrast to the above mentioned results, resting T cells were also found to negatively regulate osteoclastogenesis via production of granulocyte/monocyte colony-stimulating factor (GM-CSF) and IFN-γ by CD4 but not CD8 T cells [180]. Another in vitro study demonstrated that the downregulatory effect of lymphocytes is due to the CD8 T cell subset, and independent of IL-4 and TGF-β [181]. On the other hand, activated T cells were shown to promote osteoclastogenesis in vitro andin vivo. Activated (by anti-CD3ε and anti-CD28) CD4 T cells exerted their effect via membrane-bound and secretory RANKL. Transfer of ctla4–/– bone marrow cells in ragl–/– mice led to a significant decrease in bone mineral density. Consistent results were achieved by direct transfer of purified ctla4–/– T cells in ragl–/– and opgl–/– mice [3]. Recently, it was demonstrated that the effects of activated T cells on osteoclastogenesis depend on how they are activated [182]. Anti CD3ε- and anti CD28-Ab-activated T cells inhibited osteoclastogenesis, whereas T cells activated with staphylococcal enterotoxin A, PHA, and Con A had inconsistent effects. The osteoclastogenic effect was CD4+-dependent.
![Fig. 2. Cellular regulation of osteoclastogenesis. Osteoblasts/stromal cells are the main regulators of osteoclastogenesis. They express the cytokines RANKL, which binds to RANK on osteoclast precursors and thereby induces osteoclastogenesis, and OPG, which is able to prevent that interaction. Amongst others, TGF-β and 17-β-estradiol stimulate the production of OPG whereas 1,25(OH)2D3, PTH and PGE2 promote the production of RANKL. Upon activation via dendritic cells T-cells activate osteoclasts directly through the secretion of sRANKL. Furthermore, T cells secrete INFg, which on the one hand stimulates macrophages to produce pro-inflammatory cytokines which in turn promote RANKL expression in osteoblasts/stromal cells, and on the other hand suppresses permanent osteoclast activation by the destruction of TRAF6. Furthermore, endothelial cells have been shown to express RANKL and OPG and might therefore also participate in the regulation of osteoclastogenesis. MyP = Myeloid progenitor; OC-P = osteoclast precursor; aOC = activated osteoclast; OB/SC = osteoblast/stromal cell; MΦ = macrophage; T-C = T cell; DC = dendritic cell; EC = endothelial cell. Modified from Yasuda et al. [106].](https://karger.silverchair-cdn.com/karger/content_public/journal/iaa/143/1/10.1159_000098223/2/m_000098223_f02.jpeg?Expires=1716336786&Signature=qh2fu9UP855IXSrOuwVVy02Om7D6G-tmKQ9eU-YwXcGHkbDAayHje65DAqwZF-vv9fEiQX38iHC26XuUdPi3~Qjbt1OGVJqjzC4pEPIhg3DGqdVb~1YmFR-kw1QIgJZnCJN3TBbxGkGHpY4h30rLtpRrjqSUPwAofOak8voKqVG-iQk1FtRTb4x87MPm6bYS4FOPAWON2CP1wiNszDrYt2fCO82JrgGWXL-V9h0uf7aVys11TlREPBnTtVB5a~eoyxi1qMqiVXjzk7JYwI65FPMzAHnQozxWBH2tSpx-4z0a6iuI4t5OjFt8fiZAS3UCNawB0DnLk6Qf23yrVxMCpA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Cellular regulation of osteoclastogenesis. Osteoblasts/stromal cells are the main regulators of osteoclastogenesis. They express the cytokines RANKL, which binds to RANK on osteoclast precursors and thereby induces osteoclastogenesis, and OPG, which is able to prevent that interaction. Amongst others, TGF-β and 17-β-estradiol stimulate the production of OPG whereas 1,25(OH)2D3, PTH and PGE2 promote the production of RANKL. Upon activation via dendritic cells T-cells activate osteoclasts directly through the secretion of sRANKL. Furthermore, T cells secrete INFg, which on the one hand stimulates macrophages to produce pro-inflammatory cytokines which in turn promote RANKL expression in osteoblasts/stromal cells, and on the other hand suppresses permanent osteoclast activation by the destruction of TRAF6. Furthermore, endothelial cells have been shown to express RANKL and OPG and might therefore also participate in the regulation of osteoclastogenesis. MyP = Myeloid progenitor; OC-P = osteoclast precursor; aOC = activated osteoclast; OB/SC = osteoblast/stromal cell; MΦ = macrophage; T-C = T cell; DC = dendritic cell; EC = endothelial cell. Modified from Yasuda et al. [106].
Cellular regulation of osteoclastogenesis. Osteoblasts/stromal cells are the main regulators of osteoclastogenesis. They express the cytokines RANKL, which binds to RANK on osteoclast precursors and thereby induces osteoclastogenesis, and OPG, which is able to prevent that interaction. Amongst others, TGF-β and 17-β-estradiol stimulate the production of OPG whereas 1,25(OH)2D3, PTH and PGE2 promote the production of RANKL. Upon activation via dendritic cells T-cells activate osteoclasts directly through the secretion of sRANKL. Furthermore, T cells secrete INFg, which on the one hand stimulates macrophages to produce pro-inflammatory cytokines which in turn promote RANKL expression in osteoblasts/stromal cells, and on the other hand suppresses permanent osteoclast activation by the destruction of TRAF6. Furthermore, endothelial cells have been shown to express RANKL and OPG and might therefore also participate in the regulation of osteoclastogenesis. MyP = Myeloid progenitor; OC-P = osteoclast precursor; aOC = activated osteoclast; OB/SC = osteoblast/stromal cell; MΦ = macrophage; T-C = T cell; DC = dendritic cell; EC = endothelial cell. Modified from Yasuda et al. [106].
In mice it was shown that T cells are not absolutely required for osteoclastogenesis in a rheumatoid arthritis model, although they form an important pathologic feature in arthritic joints [183]. T cells in rheumatic joints are known to be in a kind of frustrated state, which is characterized by a downregulation of IFN-γ-production [184, 185]. Therefore it seems as if proinflammatory cytokines, which are produced by macrophages upon stimulation by T cells, act on synovial fibroblasts. These cells are the main source of RANKL responsible for osteoclast differentiation, although RANKL has also been shown to be produced by T cells. This model may be sufficient for explaining why T cells, which on the one hand produce IFN-γ, act osteoclastogenically by interacting with other cells in the rheumatoid joint.
Besides lymphocyte-osteoclast interactions there is now increasing awareness of the importance of osteoblasts in osteoimmunology. These cells seem to have antigen-presenting properties, since osteoblast cell lines were shown to express MHCII molecules besides the adhesins CD54 (ICAM-1) and CD166 (ALCAM), which were upregulated through IFN-γ, and could thus also activate T cells. Furthermore, osteoblasts were shown to express members of the toll-like receptor family, in particular TLR-4, TLR-5 and TLR-9, indicating an active role in host immune response. Pattern recognition receptors were found not only on the surface of osteoblasts but also intracellularly, as was recently reported by Marriott et al.[ 186] who were able to demonstrate the expression of the nucleotide-binding oligomerization domain proteins NOD1 and NOD2 following bacterial challenge of the cells. On the other hand, osteoblasts can produce IL-6 upon encountering T cells and stimulation by IL-17 [187]. Stanley et al. also demonstrated the ability of osteoblastic cell lines to present superantigen to T cells. This is of importance as superantigens are implicated in a variety of autoimmune conditions, such as rheumatoid arthritis.
Interactions of Bone Cells with Hematopoietic Stem Cells
Hematopoietic stem cells (HSC) are located in the bone marrow and are responsible for the continuous production of blood cells in an adult organism. Their capacity for self-renewal and their ability to differentiate into multiple cell types is strongly dependent on their surrounding microenvironment, which is also referred to as stem cell niche. There, cells produce various signaling molecules, cell adhesion molecules and components of the extracellular matrix and thereby determine the long-term repopulating ability of stem cells. Taichman and Emerson were among the first to notice that osteoblasts play a crucial role in stem cell maintenance due to an intimate cell-to-cell contact via integrins [5,6,7,8]. Another interesting observation was made in cbfa1 deficient mice, which were devoid of osteoblasts. In addition, those mice were characterized by the absence of bone marrow, although they showed normal hematopoietic development in liver and spleen until day E17.5, suggesting an important role for osteoblasts in HSC homing into the bone marrow cavity [31, 32]. Using a chimeric mouse model Kronenberg [188] was able to demonstrate the inhibition of HSC homing into the bone marrow after the deletion of the G-protein Gsα. Furthermore, he and others reported the supporting effects of osteogenic PTHs in HSC maintenance by stimulating bone lining cells to produce N-cadherin, important for stem cell attachment, and jagged-1, activating notch receptors on HSC [188, 189]. Arai et al.[ 190] identified a quiescent and anti-apoptotic subpopulation of HSC adhering to osteoblasts via the receptor tyrosine kinase Tie2 on HSCs and angiopoietin-1 on osteoblasts. Furthermore, the interaction of Tie2 with angiopoietin-1 increased the cadherin and intergrin mediated cell adhesion to osteoblasts and maintained the long-term repopulating activity of HSCs.
What Is Known about Large Animal (Porcine) Osteoimmunology?
Osteology is a fast developing branch in gerontological and rheumatological research, since postmenopausal osteoporosis as well as osteoporosis in elderly men is of great importance for individual well-being and public health. Most scientific work in medical research is performed in rodent models and hardly any experiments are conducted in larger animals such as cats, dogs or rabbits. Porcine cells should be paid more attention in osteology as porcine bone tissue is more closerly related to humans [191]. Thus, it would be of great benefit for osteological research to create a well characterized animal model of similar size and physiology to the human species. Like most other mammals the pig is a quadruped. As a consequence its skeleton is subjected to different forces when compared to humans, which is a weakness of this model from the biomechanical point of view. Nevertheless, static features of its bone apparatus, joints, muscles, and tendons exhibit more similarities to humans than do those of rodents. An additional major advantage of the porcine system over the rodent system is the opportunity to harvest a strikingly higher amount of bone marrow and blood cells per individual. Besides that, treatment of some osteopathologies in farm animals is also of economic interest. Well-fitting examples for such pathologic conditions in the porcine species are osteochondrosis dessicans and the humpback syndrome in fattening pigs. Many aspects of the etiology, pathogenesis, and treatment are still unclear. For that reason, in vitro models are still one important option to enlarge our knowledge concerning basic and therapeutic mechanisms in osteology. Since in pigs no information on the cytokine pattern of bone marrow-derived cells from healthy animals is currently available, our main interest was to collect basic data regarding the cytokine pattern of these cells cultured in and without presence of 1α,25(OH)2D3, which is known to promote osteoclastogenesis. This was considered an important issue, as bone marrow stromal cells (and lymphocytes) provide the milieu leading to increased or decreased osteoclastogenesis to a large extent by their cytokine production pattern. Since RANKL plays an important role in the generation of osteoclasts in humans and rodents [106], it was of particular interest to search for indications of the existence of a porcine homologue. At the mRNA level, cytokines with the most remarkable expression intensities were IL-1α, IL-6 and IL-8. Immunofluorescent staining with specific mAbs recognizing a panel of porcine cytokines revealed the presence of IL-1β and IL-6 in stromal cells and in osteoclasts. Since the production of IL-6 has predominantly been attributed to stromal cells [171], but was also shown to be produced by osteoclasts in our experiments, it appears that these cells could support their generation in a positive feedback fashion. Overall expression of TNF-α mRNA was low in the cultured bone marrow-derived cells. When investigating production of TNF-α at the protein level, stromal cells exhibited only faint immunofluorescent signals, but osteoclasts, in contrast, gave a bright fluorescence [192]. This is in accordance with the murine system, where osteoclasts are known to produce considerable amounts of TNF-α [193].
In the recent past, scientific interest in bone biology was focused on the RANK/RANKL/OPG osteoclastogenesis-regulatory cytokine system. We found that the porcine RANKL homologue not only was expressed in bone marrow-derived cells, but also could be detected in white blood cells by RT-PCR. The following sequencing step confirmed the PCR results by exhibiting a considerable nucleotide homology to human and murine sequences which was 79% in the case of the porcine sequence (GenBank Acc. No. AY606802) when compared to the human sequence. Then, we quantitatively analyzed RANKL production in cell culture supernatants by an ELISA system evaluated for the detection of human soluble RANKL. Indeed, RANKL was expressed to a significantly higher degree in cultures treated with 1α,25(OH)2D3 when compared to control cultures without 1α,25(OH)2D3[192]. This finding implicates that porcine RANKL is upregulated through 1α,25(OH)2D3, as is known from non-porcine systems [194], and might therefore be the cytokine with the highest osteoclastogenic activity in pigs. Furthermore, recent experiments demonstrated that porcine osteoblasts predominantly expressed soluble and membrane-bound RANKL besides other cytokines found in the murine osteoblast such as IL-1, IL-6, and TNF-α [195].
In conclusion, the cytokine pattern of the porcine system reflects those found in human and murine bone marrow cell cultures. Additionally, our findings provide strong evidence of the existence of the RANK/RANKL/OPG system in pigs. Further experiments should clarify the impact that 1α,25(OH)2D3 exerts on cytokine production by peripheral lymphocytes in pigs.
Currently, we are investigating the role of lymphocyte subsets on porcine osteoclast generation – seemingly peripheral blood mononuclear cells in general and CD4+ T cells in particular exert an osteoclastogenic effect in osteoclastogenesis – and immunophenotypic and cytokinologic properties of porcine osteoblasts.
In the near future it should be possible to use the ongoing increasing understanding of the osteoimmunological principles of porcine bone marrow-derived cells also to replace rodent models for osteological research by porcine in vivo models at a higher rate than today. Glucocorticoid-induced osteoporosis in minipigs may serve as an example of a porcine in vivo model of osteoporosis [196].
Osteoporosis: Immunologic Aspects
Osteoporosis is defined as a skeletal disorder characterized by compromised bone strength predisposing a person to an increased risk of fracture. Bone strength primarily reflects the integration of bone density and bone quality [197]. Osteoporosis is among the most important conditions associated with aging; the lifetime risk for a fragility fracture (vertebral fracture (fig. 3), distal forearm fracture, hip fracture) in a 50-year-old white US woman is approximately 40%, whereas that in a white US man is 13% [198].
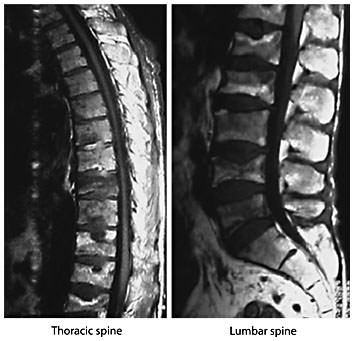
Magnetic resonance images of the thoracic and lumbar spine. Magnetic resonance study of the thoracic and lumbar spine of a man with multiple vertebral fractures due to osteoporosis. Courtesy of Prof. Dr. H. Resch, Department of Medicine 2, St. Vincent Hospital, Vienna.
Magnetic resonance images of the thoracic and lumbar spine. Magnetic resonance study of the thoracic and lumbar spine of a man with multiple vertebral fractures due to osteoporosis. Courtesy of Prof. Dr. H. Resch, Department of Medicine 2, St. Vincent Hospital, Vienna.
From a theoretical point of view, osteoporosis results from any imbalance of bone turnover that results in an excess of osteoclast activity (bone resorption) over osteoblast activity (bone formation). Nevertheless, it should be borne in mind that in an individual, bone mass is determined by the amount of bone mass achieved at skeletal maturity (‘peak bone mass’) and the velocity of subsequent bone loss. The risk of osteoporosis is strongly influenced by genetic components [199]; further determinants are hormonal and nutritional factors as well as exercise [200]. Generally, osteoporosis is classified as either primary (idiopathic) or secondary. A variety of diseases (e.g. rheumatoid arthritis [201, 202]), medications (e.g. glucocorticoids or cyclosporin A) and conditions such as alcohol abuse can result in secondary osteoporosis.
In 1998, Riggs et al.[ 203] proposed a unitary model of primary (involutional) osteoporosis in postmenopausal women and aging men. As in the model of Riggs and Melton [204], the existence of an early rapid and a late slow phase of bone loss in women is emphasized; in contrast, in aging men there is only one slow phase of continuous bone loss. The early rapid phase of bone loss in women clearly results from postmenopausal estrogen deficiency. In the late, slow phase of bone loss, vitamin D deficiency and extraskeletal consequences of estrogen deficiency lead to an increase of serum PTH levels; secondary hyperparathyroidism further stimulates bone resorption [203].
As early as 1987, Pacifici et al. [205] reported that monocytes from patients with idiopathic osteoporosis produced significantly more IL-1 than those from control subjects. In subsequent work [206], the authors demonstrated an increased IL-1 production by monocytes from postmenopausal when compared to premenopausal women or estrogen/progesterone treated women. Whereas nonosteoporotic postmenopausal women achieved premenopausal IL-1 levels within 8 years of menopause, in osteoporotic subjects an elevated IL-1 production was seen as long as 15 years after menopause. Surgically induced menopause was associated with a production of IL-1, TNF-α and GM-CSF by mononuclear cells; in women who received estrogen replacement therapy, simultaneous decreases of cytokine secretion and bone resorption were noted [207]. Whereas Zarrabeitia et al. [208] did not detect abnormalities of cytokine production in osteoporosis, Zheng et al. [209] found an increased production of IL-1, IL-6 and TNF-α by whole blood cells from patients with postmenopausal osteoporosis. Taken together these data indicate that an increased production of mononuclear cell immune products contributes to the postmenopausal enhancement of bone resorption. In this context we should like to mention that the aging process is characterized by a progressive proinflammatory status, a phenomenon referred to as ‘inflamm-aging’ by Franceschi et al. [210].
In addition to changes of cytokine production by monocytes, T cell abnormalities have been reported in patients with osteoporosis. In 1984, Fujita et al. [211] described an increased CD4+/CD8+ ratio in osteoporosis; these findings were corroborated by Imai et al. [212] and Rosen et al. [213]. Hustmyer et al. [214] described a negative correlation between the CD8+/CD56+ subset and bone mineral density. Data from our laboratory indicate that in postmenopausal women with osteoporotic fractures the CD8+/CD57+ subset is expanded; moreover, in the fracture patients the percentage of CD8+ cells that expressed TNF-α was augmented [215]. Thus, in addition to monocytes and their products, T cells appear to contribute to the pathogenesis of primary osteoporosis.
Quite surprisingly there are only few studies on RANKL or osteoprotegerin in patients with osteoporosis. Eghbali-Fatourechi et al. [216] determined the surface expression of RANKL on bone marrow mononuclear cells in premenopausal, early postmenopausal and estrogen treated postmenopausal women by flow cytometry. The surface expression of RANKL on marrow stromal cells, B cells and T cells was significantly higher in early postmenopausal when compared to premenopausal or estrogen-treated women. These findings suggest that upregulation of RANKL on stromal cells and lymphocytes in the bone marrow could mediate increased bone resorption consecutive to estrogen deficiency. Whereas the study of Eghbali-Fatourechi et al. [216] refers to the early, rapid phase of postmenopausal bone loss there are data that indicate a role of the RANKL/OPG pathway also in fracture susceptibility: Abdallah et al. [217] demonstrated an increased RANKL/OPG mRNA ratio in bone biopsies from women with hip fractures. In contrast to studies on surface expression or mRNA levels of RANKL and OPG, the measurement of these markers in serum has produced somewhat paradoxical results. With regard to OPG, most studies found elevated OPG serum levels in patients with osteoporosis [218,219,220,221] whereas one study reported decreased OPG levels in osteoporotic patients with vertebral fractures [222]. Liu et al. [223] found no differences of serum OPG and RANKL levels as well as the RANKL/OPG ratio among normal, osteopenic and osteoporotic women. Nevertheless, Schett et al. [224] showed that low levels of RANKL are a predictor of an increased risk of nontraumatic fracture. While it is possible that at least some of these observations could represent compensatory mechanisms to counteract enhanced bone degradation, the clinical utility of serum RANKL and OPG measurements still requires further investigation [225]. Further evidence for the critical role of the RANKL/OPG pathway comes from an intervention trial: the administration of denosumab, a monoclonal antibody against RANKL, in postmenopausal women with low bone mass decreased bone resorption and increased bone mineral density [226]. Thus, RANKL appears to be a promising target for the treatment of osteoporosis.
Acknowledgements
Contributing studies to this review were funded in part as Profillinienprojekt of the University of Veterinary Medicine, Vienna.
